Biolog con espectrofotometría de masas MALDI-TOF y secuenciación genética de ARN ribosómico
P.Wragg; L.Randall; A.M.Whatmore
Comparación del sistema Semi-automatizado de Biolog con espectrofotometría de masas MALDI-TOF y secuenciación genética de ARN ribosómico 16s para la identificación de bacterias de interés en veterinaria
Reseña
Los recientes avances en los métodos fenotípicos y quimiotaxonómicos han mejorado la capacidad de los sistemas para identificar bacterias a nivel de especie. La clave para el uso efectivo de estos sistemas reside en la posibilidad de recurrir a bases de datos que pueden ampliarse con nueva información sobre aislamientos atípicos o noveles. En el presente estudio, comparamos el rendimiento del sistema de identificación Biolog GEN III (de aquí en adelante, GEN III) con la espectrometría de masas mediante MALDI-TOF (desorción/ionización por láser asistida por matriz- tiempo de vuelo) y la secuenciación del gen ARNr 16S para la identificación de aislamientos de interés en veterinaria. A fin de evaluar la capacidad de los sistemas en su mayor rango de rendimiento, se emplearon las cepas más difíciles de identificar con los métodos de rutina. Durante un período de 18 meses, se analizaron 100 cepas mediante los tres métodos. Para destacar la importancia de la identificación a nivel de especie, se diseñó un sistema de puntaje ponderado capaz de diferenciar la capacidad de identificación a nivel de género y a nivel de especie. Al comparar con el estándar de referencia, el puntaje ponderado total relativo fue 0,869:0,781:0,769, mediante secuenciación del gen ARNr 16S, sistema GEN III y espectrometría de masas MALDI-TOF, respectivamente. El rendimiento a nivel de género resultó significativamente mejor utilizando la secuenciación del gen ARNr 16S. Sin embargo, el rendimiento a nivel de especie fue similar para los tres sistemas.
- Introducción
Gracias al perfeccionamiento tecnológico observado en las últimas décadas, los laboratorios de microbiología que procesan especímenes clínicos de rutina han adoptado técnicas metabólicas automatizadas o semi-automatizadas. Sin embargo, la variación intra especies y el frecuente reconocimiento de nuevas especies comenzaron a socavar la precisión de sistemas fenotípicos menos sofisticados (O’Hara, 2005). Dichos sistemas encontraron limitaciones, ya sea por incompatibilidad con grupos específicos o por falta de flexibilidad en la base de datos de referencia para reconocer perfiles nuevos y ofrecer soluciones “intuitivas”. La secuenciación del gen ARNr 16S ofreció una resolución mejorada a nivel de género y, en varios casos, a nivel de especie. De todos modos, los abordajes genómicos también presentan sus limitaciones, y la posibilidad de depositar distintas secuencias libremente en las bases de datos de acceso público, sin revisión de pares, ha puesto en riesgo la integridad de los datos y la confianza en los resultados (Mignard y Flandrois, 2006, Woo et al., 2008). La espectrometría de masas mediante desorción/ionización por láser asistida por matriz-tiempo de vuelo (MALDI-TOF) es ampliamente reconocida como uno de los métodos quimiotaxonómicos más adaptables, que puede aplicarse tanto para el diagnóstico clínico como para el campo de la investigación (Bessède et al., 2011, Hijazin et al., 2012, Bizzini y Greub, 2010). Los sistemas de espectrometría de masas MALDI-TOF existentes proporcionan extensas bases de datos y están equipados con software que puede recurrir tanto a datos locales como a datos del fabricante. El desarrollo de abordajes fenotípicos clásicos también ha continuado, aunque sin gozar de tanto reconocimiento en otras tecnologías. Biolog (Biolog Inc., Hayward, EE. UU.) adaptó el conocido principio de utilización de sustratos, combinando la actividad metabólica con la reducción simultánea de un indicador redox medida colorimétricamente dentro de una placa ELISA de 96 pocillos (Bochner, 1989, Bochner, 2008). Manifestaciones anteriores utilizaron placas específicas de acuerdo con la reacción Gram, pero, si bien resultaron exitosas, eran menos efectivas que la secuenciación de ARNr 16S para identificar “bacterias atípicas” a nivel de especie (Morgan et al., 2009). La precisión mostraba variación según los taxones; la especificidad era superior con fermentadores Gram positivos e inferior con no-fermentadores no reactivos (Holmes et al., 1994). Sin embargo, con taxones de mayor reacción bioquímica, el sistema mostró gran capacidad de identificar especies que, de otro modo, hubieran sido “difíciles de identificar” y, en algunos casos, la capacidad de detectar relaciones entre cepas epidemiológicamente asociadas (Jánosi et al., 2009, Lanka et al., 2010). En su manifestación más reciente, GEN III, se diseñó una única placa para cubrir los taxones aeróbicos gramnegativos y grampositivos, donde se observó una reducción de costos y mejoría de la flexibilidad. Este estudio fue diseñado para indicar el potencial relativo de los métodos antes mencionados como herramientas de identificación de primera línea dentro de la Agencia de Salud Animal y Laboratorios Veterinarios (AHVLA), evaluando su rendimiento en comparación con un rango de aislamientos enviados a un laboratorio especializado en bacteriología determinativa.

- Materiales y métodos
2.1. Selección y caracterización inicial de aislamientos
La mayoría de los aislamientos empleados en este estudio (n = 100) consistieron en cepas de campo derivadas de especímenes clínicos enviados al Laboratorio de Bacteriología Determinativa de AHVLA (AHVLA, Bury St Edmuns, Reino Unido), si bien también se incorporaron algunas cepas de la Colección Nacional de Cultivos Tipo (NCTC, Colindale, Londres). Los aislamientos se cultivaron en agar Columbia (Oxoid, Basingstoke, Reino Unido) complementados con un 5% de sangre de cordero e incubados de acuerdo con los requerimientos de crecimiento, ya sea de modo aeróbico o en un entorno enriquecido con dióxido de carbono (7,5% CO2), a 37 ºC durante 18 a 24 horas.
2.2. Biolog MicroStation con sistema de microplacas GEN III
En la mayoría de los casos, se seleccionó una única colonia y se la emulsionó en “fluido de inoculación A” (Biolog) para su posterior inoculación en la placa de prueba MicroPlate (Biolog). Los organismos más exigentes, incluidas las cepas capnofílicas, se cultivaron en un medio alternativo, de acuerdo con las instrucciones del fabricante, y se preparó el inóculo a una transmitancia específica, utilizando un turbidímetro, según lo detallado en la guía del usuario. Para cada aislado, se inocularon 100 μl de suspensión celular en cada pocillo de la microplaca, empleando una pipeta de múltiples canales e incubando a 37 ºC durante 20 horas, ya sea de forma aeróbica o en 7,5% de CO2, de acuerdo con las características de crecimiento. Después de transcurridas las 20 horas, se leyeron las microplacas en el lector semi-automatizado de MicroStation, y el software del sistema de identificación (base de datos GEN III, versión 5.2.1) fue el encargado de interpretar los resultados. El sistema señaló aquellos aislamientos que no pudieron identificarse después de 20 horas y que requerían mayor tiempo de incubación. Dichos aislamientos volvieron a ser incubados y leídos entre 3 y 6 horas más tarde. Aquellos que después de 26 horas no pudieron identificarse, se registraron como “sin identificación”.
2.3. Identificación mediante la secuenciación del gen ARNr 16S
Se prepararon lisados bacterianos crudos directamente a partir de las placas de cultivo, suspendiendo las bacterias de un cultivo clonal en 100 μl de agua grado PCR-RT (aproximadamente 2.0 según la escala de McFarland) y colocándolas en una placa caliente a 100 ºC durante 10 min. Se amplificó un fragmento de ~ 1400 bp de ARNr 16S de las cepas bacterianas utilizando el par de cebadores universales 27 F 5′ AGAGTTTGATCCTGGCTCAG 3′ y 1389R 5′ ACGGGCGGTGTGTACAAG 3′. Los amplicones de PCR resultantes se secuenciaron en el laboratorio con los cebadores directos e inversos 5′ GTTGCGCTCGTTGCGGGACT 3′ y 5′ CTCCTACGGGAGGCAGCAG 3′, mediante un analizador de ADN ABI 3730XL (Applied Biosystems, Warrington, Reino Unido) y métodos de secuenciación estándar. Se alinearon los datos de ambas cadenas en SeqMan (DNASTAR Lasergene 9 Suite) para generar un cóntigo de hasta 700 bp. La secuenciación se repitió o se excluyó del estudio cuando fue posible a partir de un solo cebador o cuando la longitud del cóntigo resultó menor a 500 bp posterior a su edición. Luego se utilizaron las secuencias consenso para comparar con bases de datos en línea (NCBI BLAST – http://blast.ncbi.nlm.nih.gov/Blast.cgi) y con el Proyecto de Base de Datos Ribosomal (http://rdp.cme.msu.edu/). Se aplicaron criterios de identificación de > 99% de identidad de secuencia para la identificación a nivel de especie (Drancourt et al., 2000), en donde la coincidencia debía corresponderse con la “cepas tipo” de especie. Las identidades de las cepas tipo, y los números de acceso en NCBI para secuencias de ARNr 16S equivalentes, se encuentran disponibles en http://www.bacterio.cict.fr/ para todas las especies bacterianas válidas publicadas, si bien el lector debe saber que a menudo se publican múltiples secuencias para las cepas tipo, algunas de las cuales presentan mejor calidad que las que aparecen en el enlace antes mencionado.
2.4. Espectrometría de masas mediante desorción/ionización por láser asistida por matriz- tiempo de vuelo (MALDI-TOF)
Se empleó un equipo Bruker Autoflex II (Bruker Daltonik GmbH, Bremen, Alemania) para el análisis de espectrometría de masas MALDI-TOF, siguiendo los protocolos estándar de Bruker disponibles en línea y de acuerdo con la descripción antes mencionada (Eigner et al., 2009). Los espectros desconocidos resultantes se compararon con el espectro de referencia (biblioteca de referencia de MALDI Biotyper versión 3, Bruker Daltonik) de Bruker MSP (Main Spectral Projection), empleando el software MALDI Biotyper versión 2.0.10.0 para obtener la identificación basada en un puntaje asociado al grado de coincidencia con los espectros de referencia. Para aumentar la probabilidad de obtener un buen puntaje, se identificaron los aislamientos en la placa MALDI-TOF por cuadruplicado y luego se los disparó por cuadruplicado, generando dieciséis espectros y eligiendo el mejor puntaje como resultado (los espectros replicados a partir de los puntos y disparos replicados arrojaron, en general, la misma identificación, si bien los puntajes variaron de acuerdo con la calidad del espectro individual). Los niveles de confianza para las identificaciones de espectrometría de masas MALDI-TOF fueron los asignados por el software Biotyper. Se definió que los aislamientos con un puntaje mayor a 2,000 se identificaban a nivel de especie con niveles de confianza variables, los aislamientos con puntajes de 1,7 a 1,9999 se identificaban a nivel de género únicamente y los aislamientos con puntajes menores a 1,7 no se identificaban. Sin embargo, si los puntajes de ≥ 2,000 daban lugar a más de una especie, el nivel de confianza se consideraba “de género” solamente.
- Resultados
3.1. Puntuación del resultado según su resolución a género o especie
La Tabla 1 muestra un resumen de los resultados. A fin de reflejar el valor de la identificación a nivel de especie para el laboratorio diagnóstico, se diseñó un sistema de puntaje ponderado basado en la comparación con el gold standard representado por la secuenciación del gen ARNr 16S, y denominado “identificación presuntiva” en la Tabla 1. Cuando la identificación presuntiva arrojaba un resultado indeterminado mediante secuenciación de ARNr 16S únicamente, se hacía referencia a cualquier característica inequívoca o al uso de cepas tipo para lograr una mayor determinación de las especies de la mayoría de las cepas (n = 76). Cuando la identificación presuntiva a nivel de especie no pudo ser confirmada, las cepas pudieron ser comparadas a nivel de género únicamente (n = 24). Las identificaciones a nivel de género y especie recibieron puntajes de 1 y 3, respectivamente, excepto los dos aislamientos de Salmonella, en donde el máximo puntaje alcanzado fue 1. La imposibilidad de obtener una identificación mediante cualquiera de las técnicas resultó en un puntaje de 0. El máximo puntaje teórico alcanzado para cualquier técnica, basado en la comparación con 76 cepas confirmadas a nivel de especie y 24 a nivel de género, fue de 252. Al comparar con el gold standard, el puntaje ponderado total relativo fue 0,869:0,781:0,769, mediante secuenciación de gen ARNr 16S, sistema Biolog y espectrometría de masas MALDI-TOF, respectivamente.
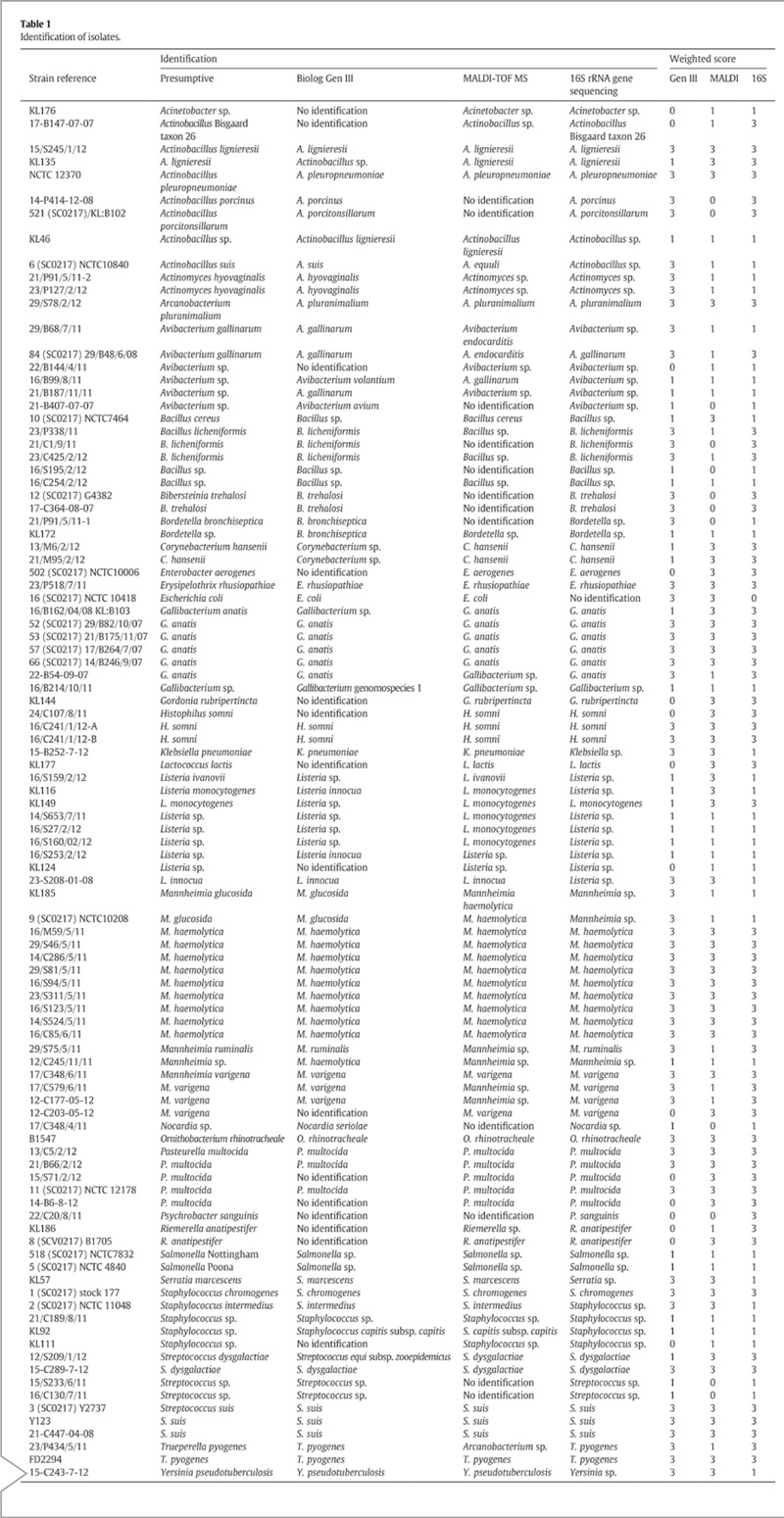
Tabla 1. Identificación de aislamientos.
3.2. Sistema de identificación Biolog GEN III
El software GEN III ofreció indicaciones claras acerca del grado de falta de certeza para la identificación a nivel de género y especie. Si bien la base de datos del fabricante representaba todas las cepas gold standard, algunas no pudieron ser identificadas a nivel de género ni de especie (n = 15). Esto se hizo especialmente evidente en los aislamientos con menor actividad metabólica, por ejemplo, Riemerella spp., en donde la segregación depende de la actividad en muy poca cantidad de pocillos. A nivel de especie, se identificaron correctamente 56 aislamientos; otros 29 fueron identificados a nivel de género, sin identificaciones erróneas, lo que arrojó una capacidad total de 85 cepas que pudieron identificarse al menos a nivel de género. El puntaje ponderado total fue de 197/252.
3.3. Espectrometría de masas MALDI-TOF
El número de cepas que no pudieron identificarse, al menos a nivel de género, fue menor que con GEN III (n = 12), mientras que el número de cepas identificadas a nivel de especie fue marginalmente menor que con GEN III (n = 53). Otras 35 cepas se identificaron correctamente en lo que respecta a género, sin identificaciones erróneas a nivel de género, dado que, al momento de realizar el estudio, el cambio de Arcanobacterium pyogenes a Treperella pyogenes aún no se había producido (Yassin et al., 2011). El número total de cepas identificables al menos a nivel de género fue de 88, con un puntaje ponderado total de 194/252.
3.4. Secuenciación de ARNr 16S
Solo una cepa no pudo ser identificada con la secuenciación de ARNr 16S (cepa 10418 de NCTC, Escherichia coli). Teniendo en cuenta los criterios estrictos empleados para la nomenclatura, se identificaron 60 cepas a nivel de especie y 39 a nivel de género. El puntaje ponderado total fue de 219/252.
3.5. Análisis estadístico
Se comparó el rendimiento total de los tres sistemas en relación con dos medidas binarias.
La primera suponía que el diagnóstico era correcto si el puntaje era de al menos 1 (identificación a nivel de género), y la segunda lo consideraba correcto solo si el puntaje de clasificación era de 3 (identificación a nivel de especie). Se empleó la prueba de McNemar para comparar la proporción de resultados correctos para cada par de sistemas. Para la identificación a nivel de género, se compararon las proporciones de muestras con un puntaje de ≥ 1 para los sistemas GEN III y espectrometría de masas MALDI-TOF, y no se encontraron diferencias significativas entre los dos sistemas (p = 0,69), lo que indica un rendimiento similar en este nivel de identificación. Además, en este sentido, la secuenciación de ARNr 16S presentó un rendimiento significativamente superior que GEN III y la espectrometría de masas MALDI-TOF (p = 0,0005 y 0,0034, respectivamente). Sin embargo, para la identificación a nivel de especie (puntajes de 3), no hubo evidencia de una diferencia significativa entre los sistemas GEN III, MALDI-TOF o la secuenciación de ARNr 16S (p = 0,7428; 0,5847 y 0,2295, respectivamente), lo que indica que las tres técnicas mostraron un rendimiento altamente similar en este nivel.
- Discusión
Los estudios comparativos acerca del rendimiento de los sistemas de identificación deberían considerar la importancia de la identificación a nivel de especie y la relevancia de la identificación errónea. A nivel clínico o epidemiológico, los datos erróneos o limitados podrían resultar en regímenes de tratamiento inadecuados o en un análisis estadístico de prevalencia engañoso. Si bien el sistema de puntaje ponderado empleado en el presente estudio destacó la importancia de la identificación a nivel de especie, no hubo intención de cuantificar los efectos negativos de la identificación errónea, por ejemplo, con la elaboración de un puntaje negativo, aunque se presentaron relativamente pocos ejemplos. Además, el objetivo primario del estudio no fue la evaluación de la repetibilidad de los métodos.
Resulta curioso que, en lugar de identificar erróneamente la especie, o peor aún, el género, GEN III tendía a arrojar el resultado de “Sin identificación”, lo que indicaba que el usuario debía buscar métodos alternativos o repetir el análisis. La falta de identificación a nivel de especie (n=2) se limitó a Listeria monocytogenes, cepa KL116, identificada erróneamente como L. innocua, y Streptococcus dysgalactiae, cepa 12/S209/01/12, identificada erróneamente como S. equi. Se observaron algunas diferencias en la capacidad del sistema para identificar todas las cepas de una especie determinada, por ejemplo, en el caso de Pasteurella multocida. Esto puede atribuirse a la variación intra-especie (cepa), aunque es probable que exista variación entre ensayos. La identificación de los aislamientos dentro de la familia Pasteurellaceae puede resultar especialmente difícil y se representaron 48 cepas en este estudio. GEN III ofreció un buen rendimiento en este área, arrojando un puntaje total de 111, sin mostrar identificaciones erróneas conocidas.
Para la espectrometría de masas MALDI-TOF, se produjeron identificaciones erróneas a nivel de especie (n = 5) por dos razones asociadas a la base de datos, ya sea la falta genuina para obtener un resultado correcto (como en el caso de dos cepas de Avibacterium gallinarum identificadas erróneamente como A. endocarditis, y una cepa de Actinobacillus suis identificada erróneamente como A. equuli) o los cambios en la antigua nomenclatura (como en el caso de dos cepas de Mannheimia glucosida, antes denominada M. haemolytica serotipo A11 e informada como M. haemolítica). La identificación dentro de Pasteurellaceae arrojó un puntaje total de 103, incluidas las identificaciones erróneas mencionadas anteriormente. La identificación de especies dentro de ciertos géneros de Pasteurellaceae se ha destacado como un área de debilidad potencial para la espectrometría de masas MALDI-TOF por parte de otros autores (Kuhnert et al., 2012). Las mejorías en las bases de datos indudablemente aumentarán el rendimiento para identificar las bacterias denominadas “difíciles de identificar” con algunos autores (Bizzini et al., 2011), con tasas de identificación en relación con el nivel de especies tan bajas como 45,9%. Veloo et al. (2011) alcanzó tasas de identificación de nivel de especie entre anaerobios desde 51 hasta 61%, dependiendo del instrumento utilizado. La necesidad de realizar pruebas específicas adicionales donde se produjeron coincidencias de especies (con puntajes de logaritmo ≥ 2.0) y un ajuste a los criterios de aceptación donde las diferencias en el puntaje de logaritmo de < 0,200 se produjeron en múltiples coincidencias llevó a algunos autores a recomendar la participación de un bacteriólogo especializado con conocimiento de taxonomía para interpretar el resultado de la espectrometría de masas MALDI-TOF (De Bel et al., 2011). Aunque la secuenciación genética de ARNr 16S alcanzó el mayor puntaje general (219/250), el análisis indicó que, si bien se obtuvieron más resultados a nivel de género, al emplear los criterios estrictos de nomenclatura aplicados aquí, los resultados no fueron mejores que los otros dos sistemas a nivel de especie, con reconocidas áreas de debilidad en ciertos géneros (por ejemplo, Listeria sp.). Dentro de Pasteurellaceae, la secuenciación de ARNr 16S alcanzó el mayor puntaje (123) sin identificaciones erróneas conocidas. La superioridad de la secuenciación de ARNr 16S por sobre los sistemas fenotípicos automatizados (Fontana et al., 2005, Bosshard et al., 2006) quizás no resulte sorprendente. Sin embargo, los estudios sugieren que, en relación con determinados géneros, la secuenciación de ARNr 16S puede ser falible a nivel de especies (Janda y Abbott, 2007, Mignard y Flandrois, 2006). Sin dudas, el valioso aporte de un bacteriólogo especializado es esencial independientemente del sistema de identificación utilizado.
Las iniciativas para el mejoramiento de las bases de datos son continuas en las tres tecnologías, tanto impulsadas por el fabricante como por los centros de investigación y grupos de interés de todo el mundo. Más importante aún, los tres métodos permiten el desarrollo de bases de datos definidas por el usuario a fin de incorporar diversidad de cepas, una característica fundamental de cualquier sistema moderno de identificación. Cualquier sistema privado de esta funcionalidad se verá rápidamente afectado por datos de mala calidad y la incapacidad de estar a la altura de los rápidos cambios que existen en la nomenclatura y el descubrimiento de “nuevas” especies. Para la secuenciación de ARNr 16S, los sistemas comerciales como MicroSEQ (Applied Biosystems, Foster City, EE. UU.), permiten la búsqueda automática de una extensa base de datos validada con la facilidad de buscar bases de datos públicas como GenBank. Al considerar el uso de una base de datos pública, la comparación con los datos de secuencia que no se hayan sometido a revisión de pares requiere una interpretación especializada y criterios claramente establecidos para la aceptabilidad de umbrales de similitud (Boudewijns et al., 2006, Janda y Abbott, 2007, Woo et al., 2009). Bruker ofrece una base de datos flexible de extensión similar, aunque la referencia a los espectros que se encuentran fuera de la base de datos del fabricante requiere complementarse con cepas validadas. La posibilidad de compartir espectros entre laboratorios ofrece una gran promesa, aunque se deberá puntualizar la consistencia en los métodos de producción. GEN III está equipado con una extensa base de datos del fabricante, que puede complementarse haciendo referencia a los datos definidos por el usuario. No se conoce el alcance en el que pueden compartirse los datos entre laboratorios.
Además de los requerimientos de bases de datos, incluida la facilidad de ampliación, la elección del sistema de identificación involucra factores como velocidad de respuesta, rendimiento, requisitos de capacitación, vinculación con sistemas de gestión de información existentes y un análisis de costos general. Se ha propuesto un umbral de 5–7000 identificaciones por año para justificar la inversión en la espectrometría de masas MALDI-TOF (Prod’hom et al., 2012). La secuenciación genética de ARNr 16S permanece limitada en su aplicación más allá de los laboratorios de referencia e investigación, debido al costo y a la necesidad de experiencia en la interpretación (Claridge, 2004). En este contexto, las opciones más asequibles, basadas en análisis fenotípico clásico, podrían elegirse en los pequeños laboratorios que no poseen los medios para justificar o realizar la compra inicial y el mantenimiento rutinario de los sistemas de espectrometría de masas MALDI-TOF o secuenciación de ARNr 16S. La ventaja significativa de la espectrometría de masas MALDI-TOF es la disponibilidad de los resultados en menor tiempo comparado con los métodos fenotípicos tradicionales (Cherkaoui et al., 2010) o la secuenciación genética de ARNr 16S, aunque esto supone que se obtenga un “valor de puntaje” suficientemente elevado, que indica el nivel de confianza requerido en el resultado, para una única especie sin necesidad de repetir las corridas o alterar la matriz de muestra.
- Conclusión
Las relativas fortalezas y debilidades demostradas en los tres sistemas destacan el aporte realizado por el bacteriólogo especializado en dos áreas clave: en primer lugar, la capacidad de seleccionar la técnica más adecuada para el aislado desconocido, basándose en el conocimiento previo de las características fundamentales del organismo (por ejemplo, historial de casos, sitio de aislamiento, y rasgos primarios como morfología general y microscópica) y, en segundo lugar, la capacidad de efectuar una referencia cruzada entre el resultado del sistema y las características fundamentales mencionadas. La primera aptitud podría considerarse un “triaging” y podría generar ahorros en el tiempo, el dinero, y potencialmente, prevenir el informe de un resultado erróneo. La segunda aptitud podría considerarse la “comprobación del resultado”. ¿El resultado del equipo se corresponde con todos los datos disponibles, incluidos el fenotipo y el historial de casos? De este modo, a pesar de que los recientes avances tecnológicos pueden indicar una disminución gradual del rol del bacteriólogo, es indiscutible que su conocimiento fundamental nunca ha sido tan importante para asegurar que los resultados de laboratorio sean de la más alta calidad.
Agradecimientos
Parte de esta investigación se presentó en el Segundo Congreso de la European Association of Veterinary Laboratory Diagnosticians (EAVLD), Kazimierz Dolny, 1–4 Julio, 2012.
Agradecemos a Therese Carson, AHVLA Bury St Edmunds y la unidad de aseguramiento de calidad de AHVLA VetQas (VetQas, Sutton Bonington, Reino Unido) por proporcionar muchas de las cepas de investigación utilizadas en este estudio, y a Robin Sayers, Specialist Scientific Support Department, AHVLA Weybridge, por el análisis estadístico de los datos. La financiación para este trabajo se obtuvo del Research and Development Internal Investment Fund de AHVLA, proyectos RD0038, RD0016 y TDP061.
Referencias
Bessède et al., 2011
-
Bessède, M. Angla-gre, Y. Delagarde, S. Sep Hieng, A. Ménard, F. MégraudMatrix-assisted laser-desorption/ionization biotyper: experience in the routine of a University hospital. Clin. Microbiol. Infect., 17 (2011), pp. 533-538