




Transferencias de métodos LC exitosas y sin estrés
Autores:
Carsten Paul, Maria Grübner, Michael Heidorn, Matthias Krajewski, Sabrina Patzelt, Thomas Piecha y Frank Steiner. Thermo Fisher Scientific, Germering, Alemania
Objetivo
Explicar en detalle los parámetros instrumentales que los usuarios de HPLC deben considerar durante la transferencia de un método de HPLC analítico entre diferentes instrumentos.
Introducción
La transferencia de procedimientos analíticos en cromatografía líquida (LC) es una tarea habitual en muchos laboratorios. Este desafío se puede clasificar en los siguientes escenarios comunes:
- Aceleración de métodos, por ejemplo de HPLC a UHPLC.
- Transferencia del método a equipos idénticos, por ejemplo en otro laboratorio.
- Transferencia del método a un instrumento no idéntico, por ejemplo, a un sistema adquirido recientemente.
Después de la comercialización de instrumentos de cromatografía líquida de ultra alto rendimiento (UHPLC) y el uso simultáneo de columna de partículas de menos de 2 µm, el escenario A se convirtió en una tarea común en muchos laboratorios. Sin embargo, hay varias publicaciones disponibles que explican los principios del escalamiento de métodos. Por lo tanto, el escenario A no se detalla aquí y se remite al lector a la literatura existente.
Para los escenarios B y C, el objetivo de dicho flujo de trabajo es “simplemente” obtener resultados equivalentes entre ambos sistemas para tener rápidamente un método operativo y reducir los esfuerzos de revalidación. Para el escenario B, la robustez del método es el foco ya que el método se transfiere entre dos sistemas idénticos. Discutir sobre los criterios para la solidez/revalidación del método no está dentro del alcance de esta publicación.
El desafío resumido en el escenario C se enfrenta a menudo al transferir métodos (validados) entre diferentes laboratorios, por ejemplo, de un laboratorio en desarrollo a un laboratorio de control de calidad o, de manera similar, de un laboratorio patrocinador a un laboratorio contratado. En este caso, es necesario considerar la influencia de los parámetros del instrumento en la separación cromatográfica para una transferencia exitosa de un método de LC desde el laboratorio de origen al de destino.
Esta revisión explica los parámetros instrumentales que se deben considerar al transferir un método LC de un sistema a otro.
Además, daremos recomendaciones sobre cómo modificar ciertos parámetros para obtener resultados equivalentes. Estas modificaciones se analizan con respecto al Capítulo General de la USP <621> Cromatografía, que describe los límites aceptados de tales modificaciones. Finalmente, brindamos orientación sobre cómo caracterizar mejor la causa raíz de los problemas comunes de transferencia de métodos. Esta revisión se centra únicamente en los parámetros del instrumento. En esta publicación no se tratan aspectos como el cumplimiento correcto de un SOP, por ejemplo, la preparación de un buffer.
Categorización de métodos (U)HPLC
La importancia de los parámetros del instrumento para una transferencia de método exitosa se hizo evidente en los últimos años. La necesidad de transferir métodos gana importancia debido a la creciente participación de laboratorios externos, como laboratorios de servicios, así como la tendencia a transferir métodos globalmente dentro de una sola empresa. En ambos casos, los instrumentos de cromatografía a menudo no eran idénticos y surgieron dificultades a la hora de reproducir los resultados del laboratorio de origen. Además, la comercialización de instrumentos UHPLC con sus características físicas significativamente alteradas enfatizó la influencia de los parámetros del instrumento en una separación específica.
El grado en que un determinado parámetro influye en la tasa de éxito de un proceso de transferencia de método depende en gran medida de la aplicación real. Dos parámetros importantes son las dimensiones de la columna utilizada (diámetro interior y tamaño de partícula) y el modo de elución. La Figura 1 muestra la importancia de las principales características del instrumento durante la transferencia del método. Para simplificar, se diferencian los escenarios UHPLC (columna de 2,1 mm de diámetro interior, partículas de < 2 µm) frente a condiciones de HPLC (columna de 4,6 mm de diámetro interior, partículas de ≥ 3 µm) y condiciones de elución isocrática versus en gradiente, como se ilustra en la Figura 1.
A partir de estas consideraciones generales, resulta obvio que el volumen de retardo del gradiente (GDV) es un parámetro importante para la transferencia de un método de elución en gradiente. De manera similar, como los caudales son generalmente más bajos para las separaciones UHPLC, la importancia de hacer coincidir el GDV del sistema de origen y el sistema receptor es mayor para las separaciones UHPLC porque pequeñas diferencias en el GDV pueden afectar dramáticamente los tiempos de retención.
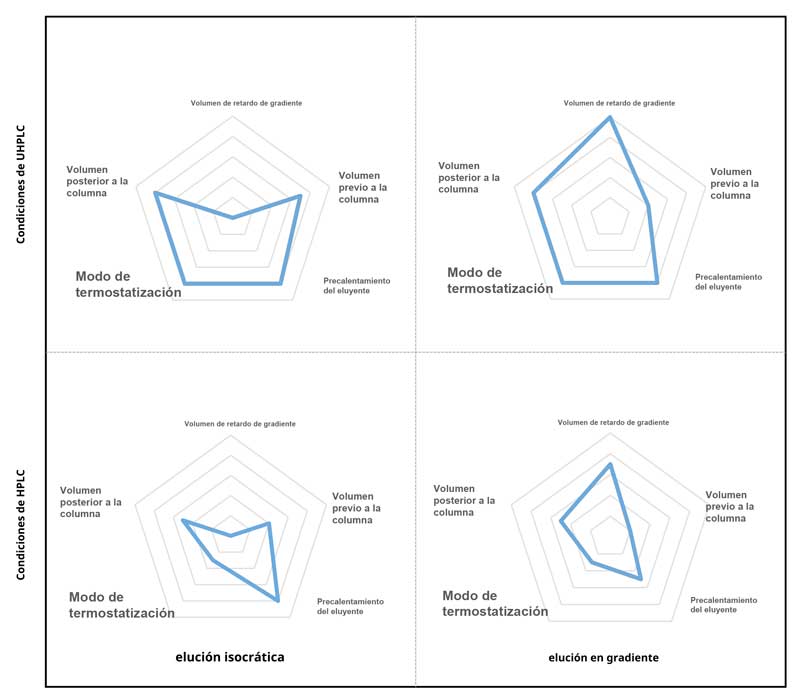
Además, es necesario considerar el modo de termostatización, que describe principalmente cómo el instrumento maneja el calentamiento por fricción dentro de la columna. Durante las separaciones por HPLC estándar, que normalmente funcionan por debajo de 400 bar (6000 psi), el calentamiento por fricción es insignificante. Por el contrario, en condiciones de UHPLC con presiones que oscilan hasta 1.500 bar (22.000 psi), se produce un calentamiento por fricción significativo. Por lo tanto, hacer coincidir los modos de termostatización es crucial al transferir métodos UHPLC.
Volumen de retardo de gradiente: ¿Qué es y cómo medirlo?
El GDV es una característica física de un sistema HPLC que describe
la capacidad de retención de todos los componentes interconectados desde el punto de mezcla hasta la entrada de la columna. Los contribuyentes al GDV pueden incluir la bomba, el muestreador automático y los capilares de conexión. Una consecuencia del GDV es que un gradiente de elución programado puede entrar en la columna con un retraso, que se puede calcular con la fórmula:
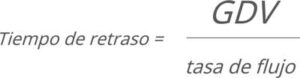
Como diferentes instrumentos de HPLC pueden tener diferentes GDV, una composición de disolvente particular puede llegar en diferentes momentos a la cabeza de una columna. Controlando el GDV puede tener un impacto dramático en la reducción de la cantidad de tiempo requerido para la transferencia de un método.
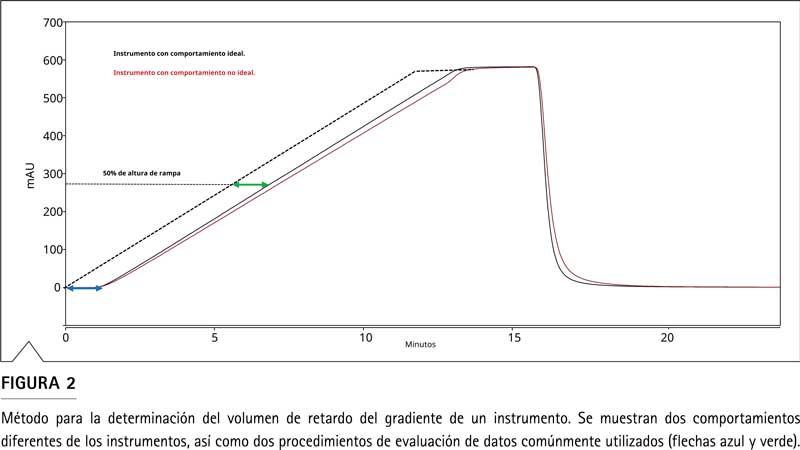
Una forma común de medir el GDV es programar un gradiente lineal de 0 % a 100 % B, con el canal B que contiene un compuesto absorbente de rayos UV. En este caso utilizamos cafeína en una concentración de 12 mg/L (Figura 2).
El GDV normalmente se calcula utilizando el momento en que la traza UV alcanza el 50% del valor máximo (flecha verde en la Figura 2) de acuerdo con la siguiente fórmula:
![]()
donde t 50% es el momento en que la traza UV alcanza el 50% de el valor máximo, tG es el tiempo total de gradiente y F es el caudal del método.
Un enfoque alternativo es utilizar la diferencia de tiempo entre el inicio del gradiente y el cruce de una extrapolación lineal de la traza UV que aumenta con la línea de base (flecha azul en la Figura 2). A partir de nuestras investigaciones, encontramos que usar el método al 50% de altura UV (flecha verde, Figura 2) es más confiable y, por lo tanto, recomendamos este enfoque. En cualquier caso, se debe tener cuidado de que no se comparen valores que provengan de diferentes métodos de evaluación.
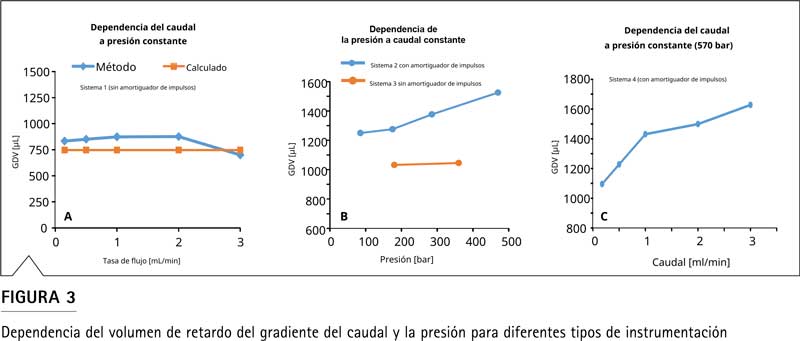
Además, el GDV no es una constante para instrumentos HPLC o UHPLC específicos, sino que depende del caudal y la presión aplicada. La Figura 3 ofrece algunos ejemplos de dependencias de caudal y presión. La Figura 3A muestra el GDV de un sistema sin amortiguador de pulso y volumen de carrera del pistón constante a diferentes caudales manteniendo constante la contrapresión del instrumento. Las diferencias entre el GDV mínimo y máximo fueron de hasta el 20 %, observándose el GDV más bajo con el caudal probado más alto de 3 ml/min. Por el contrario, la Figura 3C muestra el resultado del mismo experimento utilizando un sistema con un amortiguador de impulsos y un volumen de carrera de pistón variable. Aquí el GDV es más de un 40 % mayor con el caudal máximo de 3 ml/ min en comparación con el caudal medido más bajo. Esto sugiere que el GDV no es un parámetro instrumental fijo sino que depende del método aplicado. Para una transferencia exitosa del método, será útil determinar el GDV en las condiciones originales.
La Figura 3B muestra el efecto de la contrapresión sobre el GDV. Como era de esperar, el GDV aumenta al aumentar la presión en más del 40 % cuando se utiliza un amortiguador de impulsos. Sin embargo, a diferencia del caudal, que normalmente es constante durante una aplicación específica, la presión puede cambiar drásticamente durante la elución en gradiente. El resultado de este comportamiento es que los tiempos de retención de los compuestos que eluyen durante el gradiente se ven afectados por los GDV que cambian dinámicamente y esto debe tenerse en cuenta para una transferencia exitosa del método.
La Tabla 1 ofrece una descripción general de los sistemas HPLC comúnmente utilizados equipados con una bomba de tipo gradiente de baja presión. Como el GDV medido depende del caudal, se utilizó un caudal de 1 ml/min para todas las mediciones para garantizar las mejores comparaciones. Los sistemas que utilizan un amortiguador de impulsos dependen en gran medida de la presión de su GDV. Aunque no figura aquí, cabe señalar que el GDV de las bombas de gradiente de alta presión es generalmente menor que el de las bombas de gradiente de baja presión, lo que hace que la transferencia entre estos tipos de instrumentos sea más desafiante.
Bombas mezcladoras de baja presión versus bombas mezcladoras de alta presión
Para formar un gradiente en cromatografía líquida, existen dos tecnologías diferentes de formación de gradiente: dosificación de gradiente de baja presión (LPG) y gradiente de alta presión (HPG). En el LPG, el punto de convergencia de los disolventes (normalmente hasta 4) está antes del cabezal de la bomba mediante una válvula dosificadora de solenoide. Las bombas de LPG generalmente tienen un GDV más alto en comparación con las HPG, ya que los cabezales de la bomba contribuyen al GDV.
La diferencia en el concepto de generación de gradiente (por ejemplo, convergencia de disolvente en el lado de baja o alta presión de una bomba) también tiene consecuencias en la precisión del flujo y del gradiente, como se muestra en la Figura 5.
Se proporciona un ejemplo simulado para un gradiente programado de agua/metanol de 0% a 100% de metanol a un caudal de 1 ml/min (Figura 5A). Para un HPG, ambas bombas independientes entregan un flujo parcial según lo determinado por la composición del gradiente deseado. Por ejemplo, con una composición de 50% de metanol, ambas bombas entregarán 500 µL/min. Sin embargo, después de hacer converger ambos disolventes en el lado de alta presión de la bomba, el caudal resultante en la columna será inferior a 1 ml/min debido a la contracción del volumen de ambos disolventes. El volumen de contracción depende del disolvente y de la composición de la mezcla.
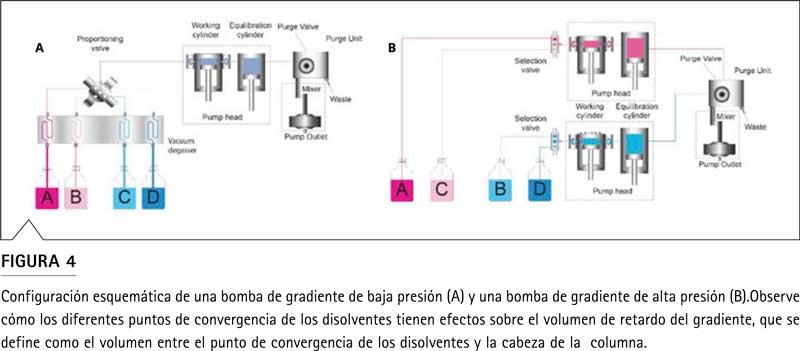
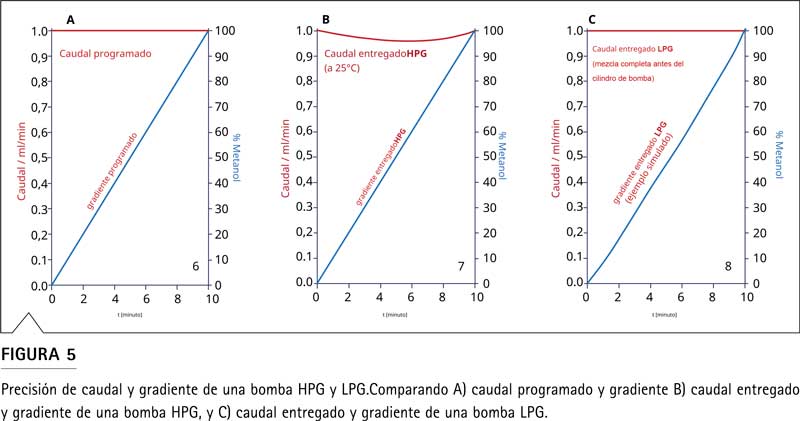
Para un gradiente de metanol, el error será de alrededor del 4% con una composición de disolvente de 55 a 60% de metanol (Figura 5B). Sin embargo, el gradiente (composición de disolvente) entregado por una bomba HPG es exactamente tan lineal como el gradiente programado (Figura 5A). La bomba de LPG, por el contrario, hace converger los disolventes antes de la bomba en el lado de baja presión y el flujo entregado en la columna será de 1 ml/min (Figura 5C). Además, debido a la contracción del volumen durante la convergencia de los disolventes en la válvula dosificadora, un LPG no suministra la composición de gradiente exacta deseada. Aquí el gradiente entregado no es lineal sino más bien curvado.
Como consecuencia de esta diferencia en el diseño de las bombas, generalmente se recomienda considerar el tipo de bomba (es decir, LPG o HPG) durante un método de transferencia del gradiente.
Preferiblemente, los métodos deben transferirse entre el mismo tipo de bomba para evitar consecuencias físicas de las diferencias de diseño que puedan obstaculizar los resultados de la transferencia de métodos. Aún así, como se describe en el siguiente capítulo, se debe tener cuidado para reflejar las posibles diferencias de GDV que pueden aparecer incluso dentro de un mismo tipo de bomba.
Ajustes de volumen de retardo de gradiente
Cuando se transfiere un método, se utilizan dos enfoques generales para adaptar los diferentes GDV de los sistemas para facilitar la transferencia del método. Nuevamente, se debe considerar que la transferencia entre HPG y los sistemas de LPG normalmente van acompañados de una diferencia significativa en el GDV y otras diferencias que hacen que la transferencia de métodos sea más desafiante. Además de los dos enfoques que se explican en las siguientes secciones, el uso de una retención isocrática al comienzo de un programa de gradiente es una práctica común en muchos laboratorios de HPLC. Cuando estos métodos se transfieren a un sistema con un GDV mayor, la retención isocrática puede simplemente acortarse. El cambio de la duración de la retención isocrática inicial está permitido según USP <621>.
Adoptando el GDV
Una forma eficaz y sencilla de compensar las diferencias de GDV entre el sistema HPLC de origen y el de recepción es cambiar físicamente el GDV del sistema receptor para que coincida con el GDV del sistema original. Una forma sencilla de cambiar el GDV es adaptar el volumen del mezclador o el volumen del bucle de muestra del instrumento al que está intentando transferir. Dichos cambios físicos del sistema se aceptan y son consistentes con las pautas de la USP.
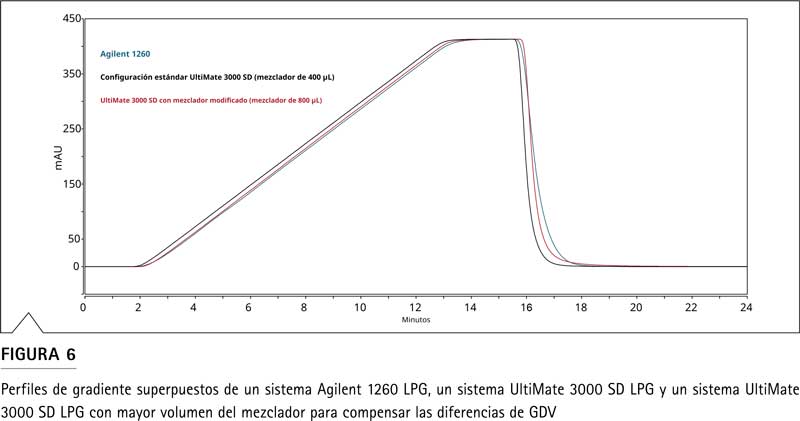
La Figura 6 ofrece un ejemplo de cómo se realizó la compensación de las diferencias de GDV para transferir un método desde un sistema Agilent® 1260 Infinity® II a un Thermo Scientific™ UltiMate™ System 3000 estándar (SD). En este caso, aumentar el volumen del mezclador de 400 µL a 800 µL en el UltiMate 3000 SD dio como resultado una buena coincidencia del perfil de gradiente.
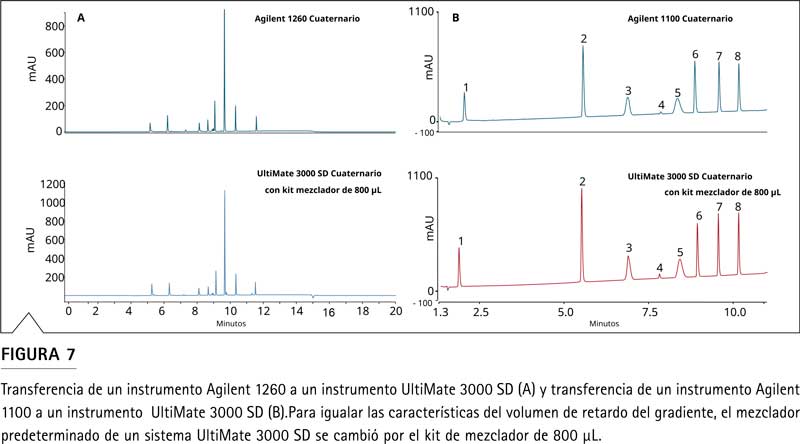
Posteriormente, se utilizó la configuración instrumental adoptada para transferir la separación de 10 pesticidas del sistema Agilent 1260 Infinity II al sistema UltiMate 3000 SD (Figura 7B). Con esta configuración, se pudo transferir el método y se logró una separación casi idéntica. También se utilizó el mismo enfoque para transferir un método para la separación de medicamentos utilizados para el tratamiento de enfermedades cardíacas de un sistema Agilent 1100 a un sistema UltiMate 3000 SD. En este caso, la instalación del kit mezclador de 800 µL también resultó exitosa (Figura 7A).
Además de cambiar el mezclador de la bomba (o el bucle de muestra en el muestreador automático), La línea de productos Thermo Scientific™ Vanquish™ UHPLC también permite el ajuste fino del GDV ajustando el GDV a través de un dispositivo de medición ubicado en el muestreador automático que contribuye al GDV del sistema. Sin embargo, como este volumen se puede ajustar con un simple comando de software, el usuario puede cambiar gradualmente el GDV para obtener el mejor método de transferencia. Con esta herramienta, es posible variar continuamente el GDV predeterminado de cualquier sistema Vanquish en un máximo de 100 µL.
Esta característica es de ayuda cuando pequeñas diferencias en el GDV impiden una transferencia exitosa del método (por ejemplo, separación a caudales de alrededor de 400 µL/min o menos o para la transferencia entre bombas binarias de bajo GDV de diferentes proveedores).
Cambiar el punto de inyección en relación con el inicio del gradiente
La segunda posibilidad de tener en cuenta diferentes GDV entre dos sistemas de HPLC es mover el punto de tiempo de inyección en relación con el inicio del gradiente. Por ejemplo, el sistema de origen podría tener un GDV de 0,8 ml y el sistema receptor un GDV de 1,8 ml, lo que daría como resultado una diferencia de 1 ml. En este caso, esta diferencia se puede compensar inyectando la muestra después del inicio del gradiente. Para un caudal de 1 ml/ min, esto significaría que la inyección se produce un minuto después de que haya comenzado el programa de gradiente. En un sentido práctico, esto significaría que el gradiente comienza en un tiempo de -1 min con respecto a la inyección, lo que siempre define el punto cero de un horario. De esta forma, la pendiente y duración del gradiente no se verían afectadas.
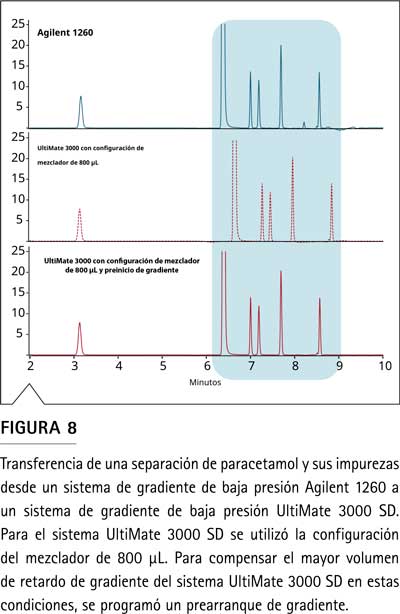
En otro ejemplo, la Figura 8 muestra la transferencia de un método para paracetamol y cinco impurezas de un instrumento Agilent 1260 a un instrumento UltiMate 3000 SD. La configuración del sistema UltiMate 3000 SD tiene un GDV predeterminado más bajo. Para compensar esta diferencia, se instaló una configuración de mezclador de 800 µL. Sin embargo, para esta aplicación que solo funciona a 120 bar, el volumen adicional del mezclador compensó en exceso la diferencia de GDV (Figura 8, cromatograma del medio). En tales casos, el software del sistema de datos de cromatografía (CDS) Thermo Scientific™ Chromeleon™ puede programar un inicio previo del gradiente para iniciar el gradiente antes del punto de inyección.. Esto resultó en una superposición perfecta de ambos cromatogramas (Figura 8, abajo), mientras que se observaron anchos de pico más pequeños para el sistema UltiMate 3000 SD.
Precalentamiento de fase móvil delante de la columna
La temperatura de un disolvente que ingresa a una columna de HPLC puede tener un impacto tanto en las formas de los picos resultantes como en los factores de retención. El preacondicionamiento adecuado de la temperatura del eluyente es esencial para lograr eficiencias óptimas de la columna, especialmente cuando se trabaja en la columna con temperaturas superiores a la del ambiente. Cuando la temperatura del disolvente entrante es significativamente menor que la temperatura de la columna, se forma un gradiente de temperatura radial entre el centro de la columna y la pared de la columna, al menos en la parte de entrada de la columna. Estas condiciones se denominan efectos de desajuste térmico y pueden tener un fuerte impacto en la forma de los picos, lo que da como resultado un ensanchamiento o una distorsión de los picos en el cromatograma. Por lo tanto, generalmente se recomienda utilizar la capacidad de precalentamiento de eluyentes de un sistema HPLC.
Para que la transferencia del método sea exitosa, se debe tener cuidado de transferir también las capacidades de precalentamiento del sistema de origen con la mayor precisión posible. Además de la simple decisión de sí o no sobre si es necesario incluir un precalentador o no, se debe considerar el diseño específico, el principio de funcionamiento y el volumen del precalentador respectivo.
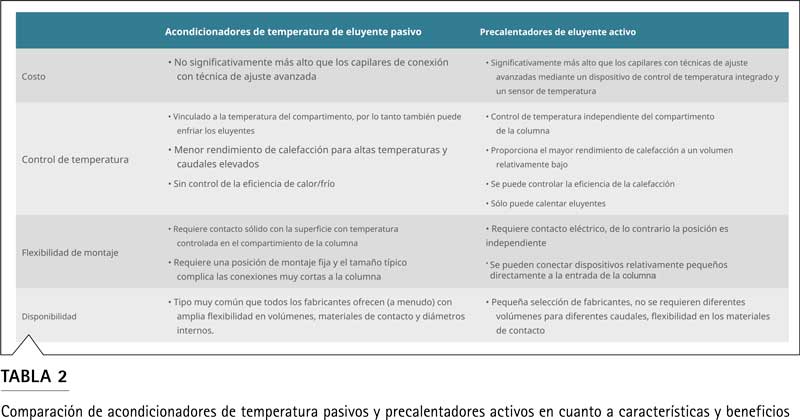
Los precalentadores activos y pasivos deben distinguirse entre dos principios funcionales fundamentalmente diferentes. Los precalentadores pasivos (o preacondicionadores de temperatura) son más comunes y funcionan según el principio de un dispositivo de intercambio de calor en contacto mecánico con una superficie de temperatura controlada en el compartimento de la columna. Desde su superficie, el calor se transfiere a través del precalentador a la fase móvil entrante a lo largo del gradiente de temperatura. Si este gradiente tiene la dirección opuesta (T Compartment < TEluent), se produce flujo de calor desde el eluyente entrante a la superficie y el dispositivo actúa como un preenfriador del eluyente. Esto se aplica cuando el compartimento de la columna se enfría por debajo de las condiciones ambientales porque el método de separación requiere bajas temperaturas. Los precalentadores activos son dispositivos que en su mayoría son independientes del control de temperatura del compartimento de la columna. Utilizan un elemento calefactor interno para regular la temperatura y controlar activamente la temperatura del eluyente resultante. El precalentador de eluyente activo de la plataforma Vanquish brinda una oportunidad única para medir y controlar la temperatura del eluyente que fluye hacia la columna, independientemente de la temperatura del compartimento de la columna. Con esto, también permite al usuario establecer la temperatura del eluyente en un valor diferente a la temperatura del compartimento de la columna, al menos dentro de ciertos límites. Mientras que los compartimentos de la columna controlan principalmente la temperatura mediante elementos Peltier que pueden calentar o enfriar según la polaridad del voltaje aplicado, los acondicionadores de eluyente activo suelen utilizar un calentador de resistencia, ya que es un dispositivo mucho menos voluminoso para montar directamente delante de la columna. La consecuencia es que sólo pueden calentar y, por tanto, no pueden acondicionarse a temperaturas inferiores a la ambiental. La Tabla 2 proporciona una descripción general de las características más importantes que distinguen a los precalentadores activos y pasivos.
Gracias al control de temperatura flexible e independiente de los precalentadores activos, proporcionan claras ventajas en escenarios de transferencia de métodos. Pueden imitar las desviaciones de la temperatura de salida esperada de los dispositivos pasivos o compensar las desviaciones en la disipación del calor por fricción de la columna. La ventaja de estas capacidades se discutirá en la sección sobre termostato de columna.
En los casos en que se utiliza un precalentador pasivo, se debe considerar el volumen, ya que normalmente es la única información disponible. En general, un precalentador con mayor volumen exhibe un efecto de precalentamiento más eficiente pero también aumenta el volumen adicional de la columna (Figura 9) y la dispersión. Esa dispersión puede ser crítica en la transferencia de métodos, especialmente para separaciones isocráticas y columnas UHPLC que generan volúmenes máximos muy bajos.

Por lo tanto, es importante hacer coincidir el volumen del precalentador con los requisitos específicos del método, teniendo en cuenta el impacto del diseño de la columna y el caudal. Se requiere la elaboración de la configuración experimental para estudiar los efectos del precalentamiento, ya que la temperatura no se puede controlar directamente con precalentadores pasivos. Los efectos del precalentamiento se investigaron con un termostato de columna de aire forzado UltiMate 3000 usando diferentes precalentadores pasivos y pasando agua a temperatura ambiente a través de una columna bajo diferentes configuraciones de temperatura elevada en el compartimiento de la columna. La temperatura de salida se registró con un sensor PT-1000 en estrecho contacto con la superficie exterior del capilar de acero inoxidable de 1/32” con aislamiento completo usando espuma tallada Styrodur™.
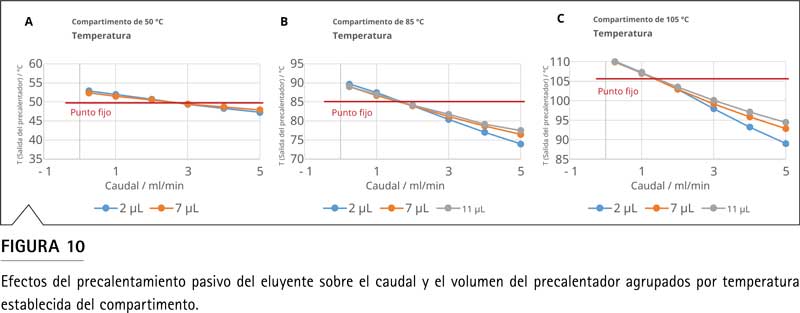
La Figura 10 muestra los resultados para temperaturas del compartimento de la columna de 50 °C, 85 °C y 105 °C con caudales de entre 0,25 ml/min y 5 ml/min y volúmenes de precalentador de 2 µl, 7 µl y 11 µl. . A la temperatura más baja, los precalentadores de 2 µL y 7 µL no fueron diferentes, por lo tanto, no se muestran los resultados del precalentador más grande. A caudales bajos, los gráficos de todas las temperaturas indican que la temperatura del eluyente saliente está por encima del punto de ajuste del compartimento de la columna. Esto elimina la idea errónea de que los precalentadores pasivos nunca pueden calentar a temperaturas superiores a las del compartimento de la columna. La razón es que la temperatura del compartimento se mide en el aire que rodea la columna y no en la placa donde está montado el precalentador. Esta placa puede estar a una temperatura más alta que el aire en el centro del compartimiento de la columna debido a la pérdida de calor durante la termostatización. Otra observación es que la pendiente creciente de la temperatura del eluyente disminuye con un mayor caudal. Estas curvas también muestran la diferenciación entre los precalentadores individuales. A medida que el volumen del precalentador aumenta y funciona a caudales muy altos, el efecto de calentamiento es mayor debido al tiempo más largo (pero aún considerablemente corto) que pasa el solvente en el dispositivo. Curiosamente, los 2 µL y las curvas de 7 µL se cruzan en todos los ajustes de temperatura.
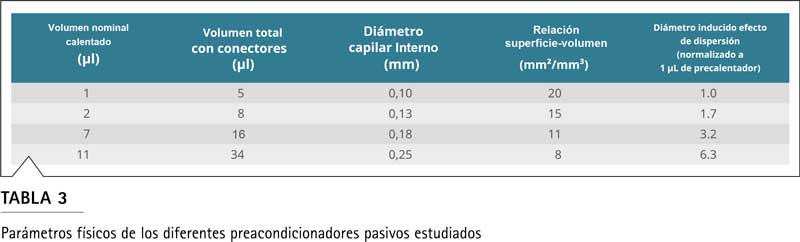
Para comprender este efecto, se deben considerar varias propiedades del precalentador (Tabla 3).
La Tabla 3 muestra que todos los dispositivos utilizados en este estudio tenían diferentes diámetros internos de capilares, lo que resultó en relaciones superficie-volumen sustancialmente diferentes. Los precalentadores de volumen más pequeño tienen relaciones superficie- volumen más altas, lo que mejoró el efecto de precalentamiento a caudales bajos cuando el tiempo que el disolvente pasa en el intercambiador de calor es suficientemente largo. La Tabla 3 también muestra el volumen total (incluido el volumen del capilar de conexión, que es sustancialmente mayor que el volumen calentado) y el diámetro interno de los precalentadores; ambos tienen un efecto pronunciado en la dispersión previa a la columna. La dispersión, que se expresa como volumen máximo resultante, disminuye con el cuadrado del diámetro del tubo (columna derecha, Tabla 3). La compensación entre calentamiento y dispersión se discutirá a continuación. De los datos de la Figura 10 se puede concluir que el precalentador de 2 µL es eficaz para caudales de hasta 2 ml/min para agua pura, que tiene una conductividad térmica notablemente mayor (factor 3 a 25 °C) que el metanol y acetonitrilo.
Los efectos combinados de la dispersión y la efectividad del calentamiento del eluyente de diferentes precalentadores pasivos se pueden ver en los cromatogramas de la Figura 11 y la Figura 12. Los cromatogramas negros en la parte superior muestran los resultados sin precalentador. La columna de 2,1 mm operada a 1 ml/min solo muestra picos ensanchados (Figura 11), mientras que el desajuste térmico en la columna de 4,6 mm provoca una división grave del pico o la formación de hombros, que aumenta con el factor de retención (Figura 12). Esto se debe al precalentamiento menos efectivo en el capilar de conexión con caudales elevados y al gradiente de temperatura radial más amplio en una columna de mayor diámetro.
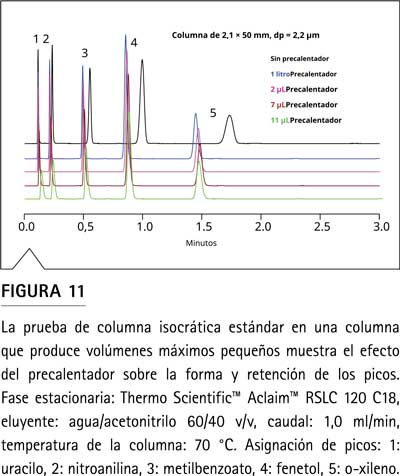
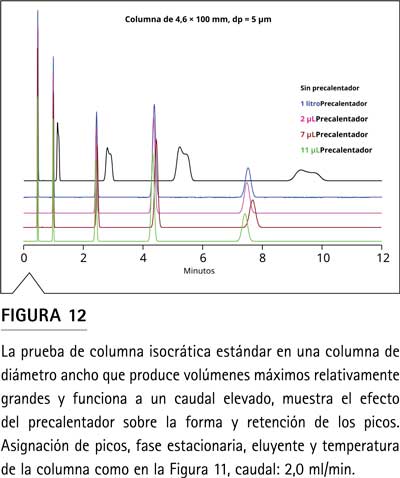
Tan pronto como se utiliza un precalentador, los picos se vuelven mucho más nítidos y el factor de retención se reduce constantemente. Estos efectos son más pronunciados en la columna de diámetro ancho y resultan de la reducción del desajuste térmico y la mayor temperatura promedio dentro de la columna cuando se usa un precalentador.
Además, las diferentes geometrías del precalentador tienen un efecto tanto en la retención como en las formas de los picos que varía mucho según la dimensión de la columna. Mientras que los primeros picos de elución se vuelven anchos y asimétricos con la columna de 2,1 mm, no hay ningún efecto negativo en la forma de los picos con la columna convencional de 4,6 mm. También es interesante ver cómo cambia la retención entre los diferentes precalentadores. Para ambos métodos, el precalentador de 7 µL produce una temperatura interna más baja que el precalentador de 2 µL, que está en línea con los datos para un caudal de 1 ml/min (Figura 9). Cuando se aplica el precalentador de 11 µL a la columna de 4,6 mm, se produce una separación con una elución de compuestos más temprana que el precalentador de 7 µL. Se podría esperar esto con temperaturas de columna más altas, pero se debe al mayor tiempo de permanencia en un precalentador con una relación superficie-volumen más similar. Con los experimentos de medición de la temperatura de salida del precalentador aplicando agua pura como fase móvil, esto fue a F = 2 ml/min y solo se observó para T = 105 °C (Figura 9). El acetonitrilo en la fase móvil de los experimentos cromatográficos conduce menos calor, por lo que las condiciones de precalentamiento serán diferentes en relación con los experimentos con agua.
Los conocimientos más profundos sobre el efecto del precalentador en la forma de los picos se pueden obtener trazando el número de platos determinado (N) de todos los picos frente a su factor de retención (k). La Figura 13 compara las curvas con y sin precalentadores en dos columnas y métodos diferentes. Mientras que el efecto del desajuste térmico se expresa como una reducción en el número de platos al aumentar la retención, el efecto de la dispersión extracolumna tiene la característica opuesta. El gráfico N vs k se puede utilizar para caracterizar si el grado de dispersión extracolumna de un sistema es apropiado para una determinada columna y método. Se puede tolerar una menor dispersión extracolumna con volúmenes de pico más pequeños, en particular para picos de elución temprana en métodos isocráticos.
Una regla básica exige que el 80 % de la eficiencia máxima que ofrece una columna en un método determinado se alcance con un factor de retención superior a 2. Sin embargo, si el número de platos disminuye en un método con retención creciente, se indica un efecto de desajuste térmico.
Aunque es difícil discriminar ambos efectos que ocurren simultáneamente, los gráficos N vs k pueden dar pistas valiosas. Las curvas para columnas UHPLC de pequeño diámetro se muestran en la Figura 13A. La operación sin precalentador (azul) muestra una menor eficiencia al aumentar la retención, lo que indica claramente un desajuste térmico. La curva para el precalentador de 1 µL (naranja) muestra una característica normal de aumento del número de platos con el segundo pico en k=3,3 que muestra el 85 % (6700) del número máximo de platos de 7900 que es aceptable. La curva para el precalentador de 11 µL (gris), comienza con una eficiencia extremadamente baja, mientras que el segundo pico en k=2,7 solo muestra el 37% (2200) de la eficiencia máxima de 5900 platos, que está muy por debajo de las 8000 placas que esta columna deberá proporcionar en el método respectivo. La Figura 13B muestra los mismos escenarios para la columna convencional de 4,6 mm. Los números de platos sin el precalentador se incluyen para que estén completos, pero se calculan a partir de picos divididos con alta retención y, por lo tanto, no son significativos. La curva para el precalentador de 1 µL (naranja) muestra una disminución lineal en la eficiencia al aumentar la retención, lo que indica un efecto de desajuste térmico. Al observar la curva del precalentador de 11 µL (gris), se puede ver un comportamiento normal para una coincidencia ideal entre columna y sistema. Hay un ligero efecto de dispersión extracolumna, que aumenta el número de placas de 8400 a 9400 entre el primer y el segundo pico retenido. Después de eso, hay una ligera disminución en el número de platos cuando se pasa a una retención muy alta. Este efecto no es un desajuste térmico, sino que resulta de una contribución más fuerte de la transferencia de masa obstaculizada expresada como un término C creciente en la ecuación de van Deemter o Knox con una mayor retención. Este efecto de transferencia de masa está presente en todos los escenarios y está más o menos oculto por el desajuste térmico o efecto de dispersión extracolumna. De la similitud de la curva naranja y gris de la columna de 4,6 mm y de las eficiencias generalmente buenas con el precalentador de 1 µL, se puede deducir que la discrepancia térmica con la combinación de precalentador pequeño y columna grande no es demasiado grave. , mientras que la ventaja de rendimiento del precalentador de 11 µL es mínima. En otras palabras, todavía sería posible utilizar el precalentador de 1 µL para la columna convencional, pero la columna UHPLC definitivamente requiere un precalentador de pequeño volumen que mantenga la dispersión fuera de la columna lo más baja posible.
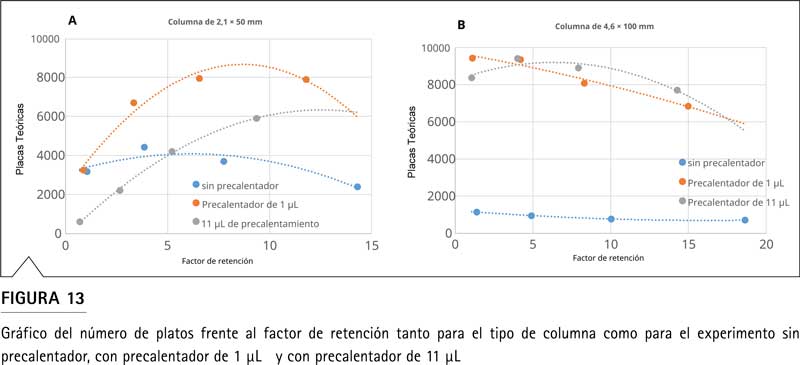
La conclusión para la selección adecuada de un precalentador pasivo en la transferencia de métodos no es fácil ni directa. Se podría demostrar la regla simple de aumentar el volumen del precalentador con el volumen de la columna, pero con precalentadores de pequeño volumen altamente efectivos en la transferencia de calor, la necesidad de aumentar el volumen del precalentador no siempre es tan fuerte, al menos mientras los caudales no excedan un cierto límite. Siempre será difícil hacer predicciones sobre el volumen del precalentador que mejor se adapte al comportamiento del sistema original, pero es ventajoso tener una variedad de dispositivos para encontrar experimentalmente el mejor. En general, siempre se debe utilizar un precalentador adecuado cuando la temperatura de la columna sea 10 °C o más por encima de la temperatura ambiente. Si existe la posibilidad de elegir, siempre se debe empezar con el precalentador más pequeño disponible. Si el efecto de calentamiento no es suficiente, esto se detectará por la mala eficiencia de los picos con mayor retención y entonces se deberá probar el siguiente precalentador más grande.
Termostato de columna y ventajas de los precalentadores activos
Los efectos de la termostatización de la columna (incluso más allá del control correcto de la temperatura en el compartimento de la columna) normalmente no se consideran en un escenario de transferencia de métodos de HPLC o UHPLC cuando se trata del análisis de la causa raíz de los cromatogramas desviados. Por ejemplo, si los tiempos de retención varían entre el sistema de origen y el de recepción, las diferencias en el GDV o el comportamiento de eliminación a menudo se consideran la única razón del efecto observado. De manera similar, si se observan diferencias en las formas de los picos, el principal problema se considera un efecto del volumen extra-columna. Sin embargo, existen diferentes modos de termostato de columna aplicados a los instrumentos de HPLC que pueden tener un efecto significativo en el cromatograma, especialmente cuando se trabaja a presiones superiores a 400 bar (6000 psi). Para aplicaciones por encima de 400 bar (6000 psi), los dos modos de termostato, aire forzado y quieto, afectarán el calentamiento por fricción producido de manera diferente (Figura 14).
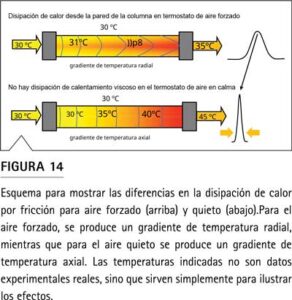
En el aire forzado se elimina más calor por fricción, lo que provoca un gradiente de temperatura radial. Por el contrario, en la termostatización con aire quieto, el calor de fricción no se elimina, lo que provoca una temperatura de separación general más alta. La retención depende de la temperatura de separación ya que la retención disminuye al aumentar la temperatura; el alcance de este comportamiento es específico de la sustancia. En tal caso, la temperatura efectiva de la columna también influye en la selectividad o la distancia de los picos.
Este efecto se ilustra con una separación de conservantes donde la selectividad del par de picos críticos (dimetilfatalato/metilparabeno) reacciona fuertemente a los cambios de temperatura de la columna. Además, el método produce calor por fricción relevante a una presión superior a 700 bar (10 000 psi), por lo que se puede esperar una fuerte influencia en el modo de termostatización de la columna (o cantidad de disipación de calor).
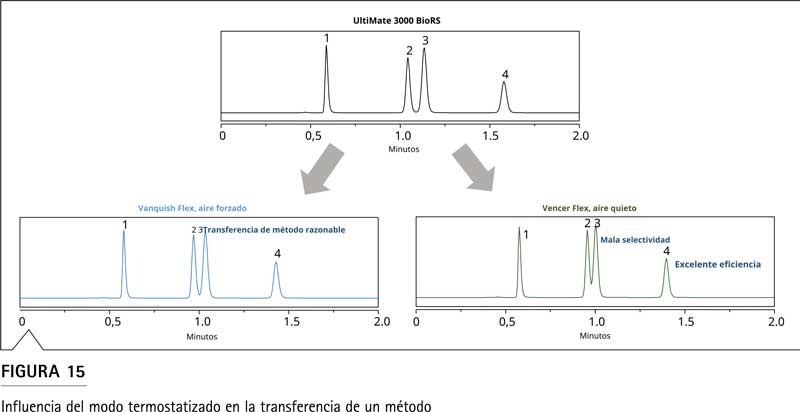
La Figura 15 muestra este efecto en el contexto de la transferencia del método isocrático respectivo de un sistema UltiMate 3000 BioRS (arriba), que emplea un principio de termostato de columna de aire forzado y precalentamiento pasivo del eluyente, a un sistema Vanquish Flex operado con aire forzado ( abajo a la izquierda) o modo de termostato de aire quieto (abajo a la derecha) con un precalentador activo. En el modo de aire forzado, el sistema Vanquish Flex permite la transferencia de métodos con una resolución aceptable del par de picos críticos. Aún así, los factores de retención de los picos 2, 3 y 4 son algo reducidos y también lo es la distancia de los picos 2 y 3. Estas diferencias surgen del hecho de que el rendimiento del UltiMate 3000 TCC y del Vanquish TCC no resulta exactamente equivalente. precalentamiento del eluyente y disipación de temperatura en sus compartimentos. Sin embargo, el modo con aire quieto no permite la transferencia del método con una separación suficiente de los picos 2 y 3 a pesar de la mejor eficiencia general de los picos.
La razón es que la temperatura global más alta en la columna, resultante del calentamiento por fricción, reduce sustancialmente la selectividad entre ftalato de dimetilo y etilparabeno. Sería deseable aprovechar la eficiencia de la termostatización de aire quieto combinada con la mejor selectividad de la temperatura más baja de la columna con la termostatización de aire forzado.
Para influir en la temperatura en la columna y, por tanto, en los factores de retención, se puede aprovechar un precalentador activo controlable independientemente y ajustado a diferentes temperaturas. Para probar esto, se realizó una serie de separaciones a partir de temperaturas iguales (40 °C) en el compartimento de la columna y el precalentador activo. La temperatura del precalentador activo se redujo gradualmente de 40 °C a 30 °C en pasos de 1 °C mientras se mantenía la temperatura del compartimento de la columna constante a 40°C.
Para demostrar el efecto, los factores de retención resultantes se correlacionaron con la temperatura del precalentador activo (Figura 16).
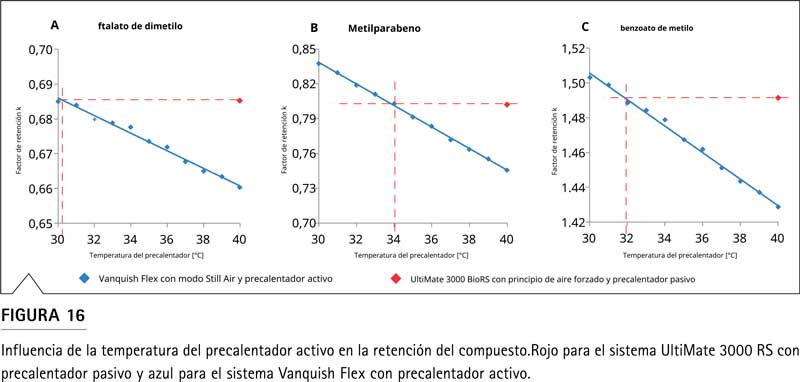
El factor de retención de ftalato de dimetilo en el sistema UltiMate 3000 BioRS se muestra como un punto rojo en el gráfico a 40 °C con un valor de 0,685 (Figura 16A). Los factores de retención en el sistema Vanquish Flex se representan como puntos azules para las diferentes temperaturas del precalentador activo. Al trazar estas dos series en un gráfico, se puede determinar la intersección de los datos rojo y azul en el eje y para comparar el factor de retención en el sistema Vanquish Flex en modo de aire quieto con el factor de retención en el sistema UltiMate 3000 BioRS. La intersección también puede indicar la temperatura correspondiente del precalentador activo, en el eje x, lo que en este caso determina que una temperatura del precalentador activo de 30,5 °C conduce a factores de retención coincidentes entre los dos sistemas para el ftalato de dimetilo.
Si se aplica este procedimiento al metilparabeno y al metilbenzoato en consecuencia (consulte otros gráficos en la Figura 16), se puede encontrar la temperatura activa del precalentador correspondiente a los factores de retención coincidentes para el metilparabeno a 34 °C y el metilbenzoato a 32 °C. Dado que los compuestos requieren tres temperaturas diferentes del eluyente entrante para igualar el factor de retención, se podría tomar el promedio de 32 °C como un compromiso para igualar los tres factores de retención lo más cerca posible.
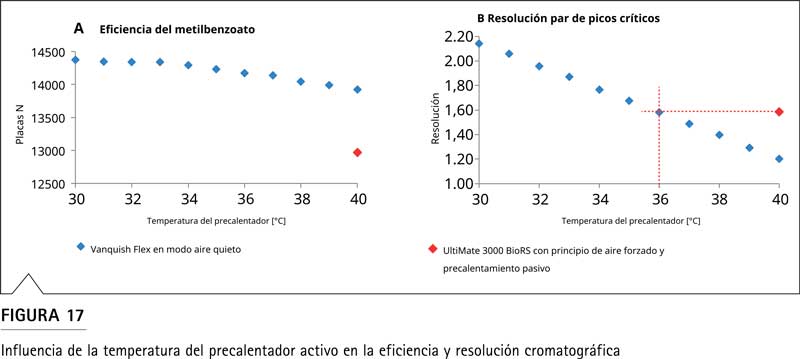
Como se indicó anteriormente, uno puede beneficiarse de los efectos positivos del modo de aire quieto bajo calentamiento por fricción a presiones de sistema más altas. Los criterios clave para esta separación son la resolución del par crítico y la eficiencia máxima general que se traduce en una relación señal/ruido mejorada en el detector. Para mostrar los efectos, la mejora de la eficiencia del benzoato de metilo en modo con aire quieto se representa gráficamente en función de la temperatura establecida en el precalentador activo. En la Figura 17A, se puede ver claramente el aumento de la eficiencia del 8% a 40 °C asociado con la termostatización del aire en calma en el sistema Vanquish Flex. El punto rojo representa el resultado en el sistema UltiMate 3000 BioRS y los puntos azules representan el resultado en el sistema Vanquish Flex en modo de aire quieto con temperatura de precalentador variable. Al reducir la temperatura del precalentador activo, no solo afecta los factores de retención sino que también puede aumentar la eficiencia, en este caso en un 10%. La razón es una compensación de un desajuste menor de temperatura radial dentro de la columna debido al flujo de calor residual (tenga en cuenta que el aire quieto no es exactamente adiabático), pero esto es solo una parte de la historia. Con esta aplicación, hay un par de picos críticos que tenían una resolución mucho peor en el sistema Vanquish Flex en modo de aire quieto que en el sistema UltiMate 3000 BioRS. Debido a la influencia de los factores de retención al disminuir la temperatura activa del precalentador, la resolución del par de picos críticos cambia. Para demostrar esto, la resolución se traza en función de la temperatura activa del precalentador, y la intersección entre la línea de puntos roja y los puntos de datos azules del sistema UltiMate 3000 BioRS y el sistema Vanquish Flex, respectivamente, muestran el punto de ajuste para el precalentador activo debe estar a 36 °C. Si bien la resolución es equivalente a la del sistema UltiMate 3000 BioRS en estas condiciones, los factores de retención no coinciden como se muestra antes. Si se observa la temperatura activa del precalentador previamente determinada de 32 °C (igualdad de retención), la resolución del par de picos críticos en el sistema Vanquish Flex excede claramente el valor observado en el sistema UltiMate 3000 BioRS.
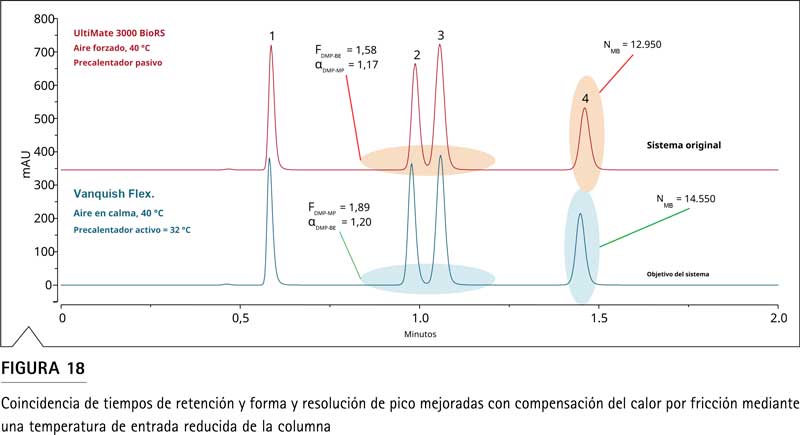
La Figura 18 compara el punto de partida en el sistema UltiMate 3000 BioRS a 40 °C y las condiciones optimizadas para el análisis en el sistema Vanquish Flex, con el compartimiento de la columna en modo de aire quieto a 40 °C y el precalentador activo configurado en 32 °C (valores de ajuste obtenidos de las evaluaciones anteriores).
Al reducir la temperatura del precalentador activo a 32 °C mientras se mantiene la temperatura del compartimento de la columna a 40 °C, se pueden igualar los factores de retención de los compuestos separados del sistema UltiMate 3000 BioRS con el sistema Vanquish Flex. Estos parámetros en el sistema Vanquish Flex también superan la resolución del valor inicial de 1,58 a 1,93 y aumentan la eficiencia en un 11,5%. Este ejemplo muestra el efecto positivo de esta propiedad única de los precalentadores activos. En condiciones de calentamiento por fricción, los precalentadores activos pueden facilitar la transferencia entre diferentes modos de termostato, incluso sin cambiar la temperatura controlada del compartimento de la columna, lo cual es difícil en un ambiente regulado.
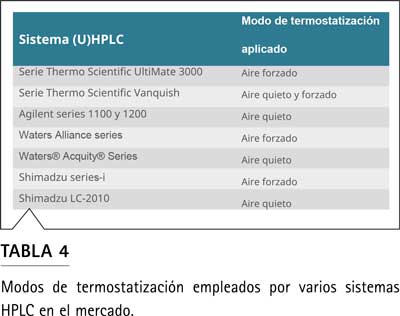
La Tabla 4 proporciona una descripción general de los modos de termostato de columna de los sistemas (U)HPLC de uso común.
Efecto del volumen extracolumna
El volumen extracolumna (ECV) es el volumen desde el inyector hasta el detector excluyendo el volumen de la columna. El ECV se puede clasificar además en volumen previo a la columna y volumen posterior a la columna. El volumen previo a la columna está determinado principalmente por las piezas del instrumento, como el asiento de la aguja y los tubos de conexión, mientras que el volumen posterior a la columna también deriva del tubo de conexión al detector y de los capilares dentro del detector, pero principalmente del volumen de la celda de flujo del detector.
El impacto del ECV en la tasa de éxito de la transferencia del método depende en gran medida del método en sí. En general, la influencia del ECV se vuelve más prominente si el volumen de la columna disminuye. Este efecto se informó para dos formatos de columna en condiciones de elución isocrática: agregar 15 μL de ECV adicionales a un sistema con una columna de 4,6 × 150 mm dio como resultado una pequeña pérdida del 1 % en la resolución para un compuesto de baja retención (k = 1) y ninguna pérdida. de resolución para un compuesto más retenido (k=5). Por el contrario, para el formato de columna más desafiante de 2,1 × 150 mm, la pérdida de resolución fue del 19% y el 3%, respectivamente, para los dos compuestos.7 Por lo tanto, una variación del instrumento en el ECV tiene una relevancia limitada cuando se trabaja con HPLC estándar. columnas. Si las columnas de 2,1 mm d.i. (condiciones UHPLC) no se puede ignorar el efecto del ECV.
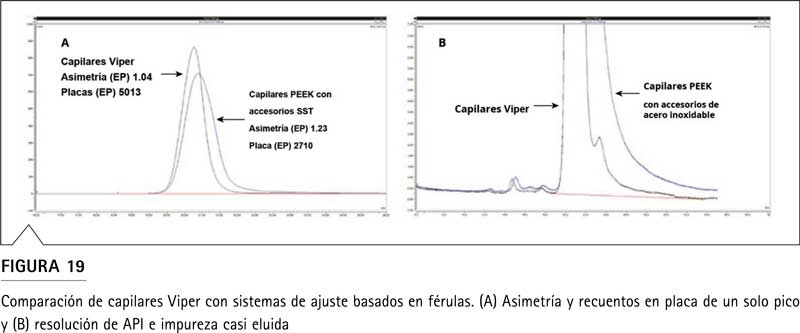
La Figura 19 muestra el impacto potencial de ECV adicional, generado por diferentes diseños de tubos, en una separación cromatográfica. La Figura 19B ofrece un ejemplo cromatográfico en el que, debido al ECV extendido, no se resolvió una impureza del pico principal, mientras que al utilizar los capilares Thermo Scientific™ Viper™ Fingertight y su ECV minimizado, la impureza se distinguía del compuesto principal. Estos efectos serán más pronunciados en las columnas de diámetro bajo que en las columnas de HPLC estándar (4,6 mm de d.i.). Por lo tanto, se debe tener cuidado con las conexiones fluídicas cuando se trabaja con columnas de 2,1 mm de d.i. o más pequeño.
Un ECV significativamente más bajo en la unidad receptora que en la unidad de origen tiene efectos perjudiciales en la separación de sustancias que eluyen tempranamente cuando se utilizan disolventes de muestra fuertes.
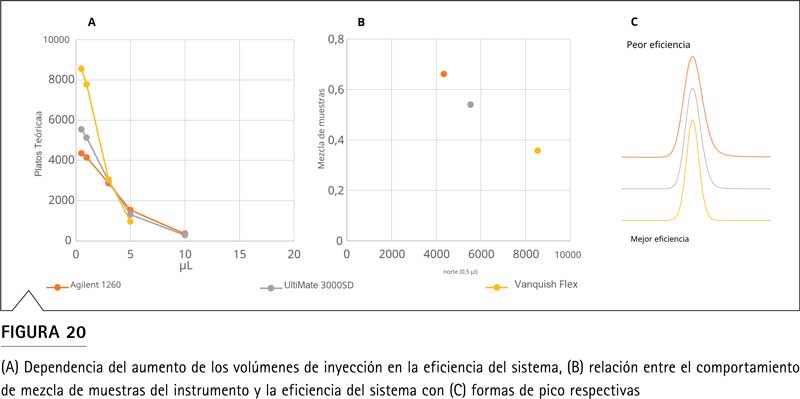
Para ilustrar este comportamiento, se utilizó una separación isocrática en condiciones de discordancia de disolventes (muestra en metanol al 100 % con condiciones de elución de agua/acetonitrilo 50:50). La Figura 20A muestra los recuentos de placas para tres sistemas diferentes frente al volumen de inyección. El sistema Vanquish Flex muestra claramente la mayor eficiencia cromatográfica para los volúmenes de inyección más bajos de 0,5 µL y 1 µL, mientras que a 3 µL o más no se observó diferencia. Además, el comportamiento de mezcla de la muestra se investigó calculando un factor de mezcla de la muestra (dividiendo el número de platos con un volumen de inyección de 3 µl por el número de platos con un volumen de inyección de 0,5 µl).
En la Figura 20B se representa el factor de mezcla para los tres instrumentos frente al número de platos con un volumen de inyección de 0,5 µL y se hace evidente una correlación. Debido a la menor eficiencia cromatográfica general, el sistema Agilent 1260 presenta una mejor mezcla de muestras antes de la columna en comparación con otros sistemas. En este caso, puede tener sentido aumentar artificialmente el volumen de precolumna, disminuir el volumen de inyección o intentar hacer coincidir el disolvente de la muestra con el eluyente para transferir un método de un sistema con mayor volumen de precolumna a un sistema con Volumen precolumna más bajo.
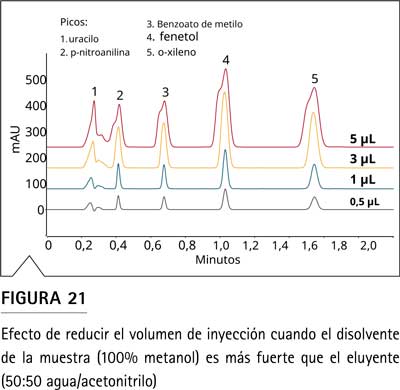
En la Figura 21, se muestra el enfoque de reducir el volumen de inyección para obtener una forma de pico satisfactoria. El volumen de inyección se puede ajustar según USP <621> si cumple con los límites de precisión y detección requeridos.
Para separaciones en gradiente, la influencia de la ECV es menor debido al efecto de reenfoque del pico en la cabecera de la columna. Además, el volumen posterior a la columna es más relevante que el volumen anterior a la columna, debido al enfoque del pico en la columna en el modo de gradiente. Aún así, las malas conexiones de fluidos, así como las dimensiones inapropiadas de la celda de flujo, pueden dar como resultado una resolución máxima diferente entre el sistema de origen y el sistema receptor al transferir un método (Figura 19).
Detector: celdas de flujo y configuración del detector
Es fundamental tener en cuenta la celda de flujo del detector al transferir métodos entre diferentes sistemas (U)HPLC. Es necesario tener cuidado de que el volumen de la celda de flujo esté de acuerdo con el volumen máximo y con el diámetro de la columna. Como regla general, el volumen de la celda de flujo no debe ser mayor que el 10 % del volumen del pico más pequeño. Si la relación entre el volumen máximo y el volumen de la celda de flujo disminuye, la consecuencia será la dispersión del pico, incluida una pérdida de eficiencia y de relación señal-ruido.
Las separaciones que se muestran en la Figura 22 se realizaron en una columna de 1,0 × 100 mm, 2,1 × 100 mm y 3,0 × 100 mm. respectivamente. Para todas las separaciones se utilizó un monitor UV de baja dispersión seguido de una celda de flujo de alta sensibilidad, con un volumen de celda de flujo iluminada de 13 µl y un recorrido de luz de 60 mm. Además, el factor de ensanchamiento de pico se calculó dividiendo el volumen de pico medido en la celda de flujo de 13 µL por el volumen de pico medido con el monitor UV. A partir de estos datos, resulta obvio que solo se observa una pérdida marginal de resolución entre la celda de flujo de 45 nL y 13 µL para la columna de 3,0 × 100 mm con volúmenes máximos entre 27 y 129 µL. Para el último pico de elución en la columna de 3,0 × 100 mm, casi no se observa ensanchamiento del pico. En este caso, la relación entre el volumen pico y el volumen de la celda de flujo es exactamente 10. Para otros formatos de columna, la celda de flujo de 60 mm de alta sensibilidad no es adecuada. Sin embargo, durante un escenario típico de transferencia de método, puede resultar poco realista que se cambie el formato de la columna. Aún así, el mismo principio (volumen de la celda de flujo 10 % del volumen máximo) se aplica a escenarios de transferencia de métodos donde el formato de la columna se mantiene constante, pero el volumen de la celda de flujo varía según se utilizan diferentes instrumentos.
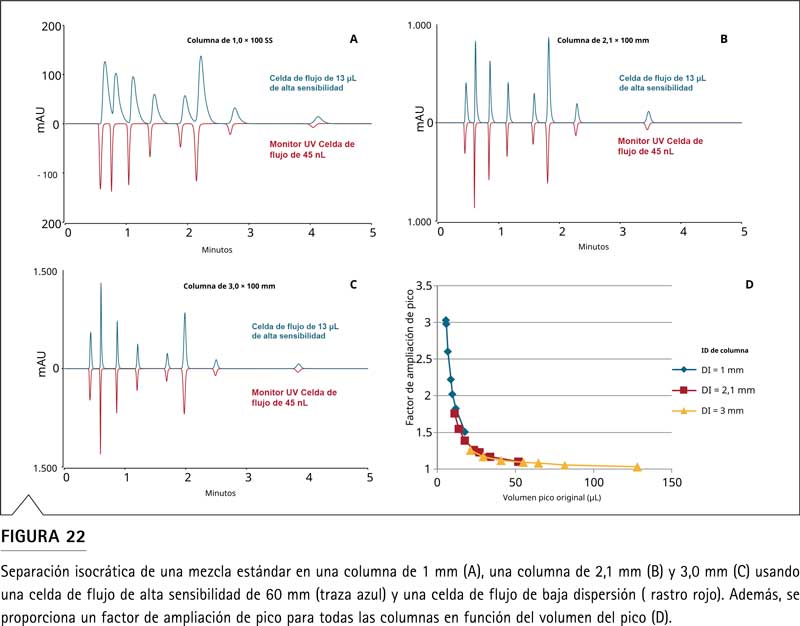
Además de las dimensiones físicas del detector, o específicamente de la celda de flujo del detector, la configuración del detector juega un papel importante en la obtención de resultados similares entre diferentes tipos de detectores o entre diferentes proveedores. Para una transferencia de método exitosa, la configuración del ancho de banda, la longitud de onda de referencia y el tiempo de respuesta son importantes. El tiempo de respuesta (también tiempo de subida o constante de tiempo) es, en general, una medida de la rapidez con la que responde el detector a un cambio de señal. Un tiempo de respuesta cada vez mayor reduce el ruido de la señal pero al mismo tiempo puede disminuir la altura de la señal y, en consecuencia, influir en la sensibilidad. Además, un tiempo de respuesta cada vez mayor aumenta la anchura del pico y desplaza el pico hacia tiempos de retención más altos.
La Figura 23 muestra el efecto en un ejemplo práctico de una cromatografía de exclusión molecular (SEC) de un estándar comercial. En este caso se observó una disminución de los platos teóricos de casi un 13%. Esto es especialmente crítico para SEC, ya que a menudo no es fácil lograr la separación inicial entre agregados de bioterapéuticos. Además, el ruido se reduce drásticamente para lograr un mayor tiempo de respuesta y mejora la relación señal/ruido en general, por lo que el usuario debe encontrar un compromiso para obtener los mejores resultados.
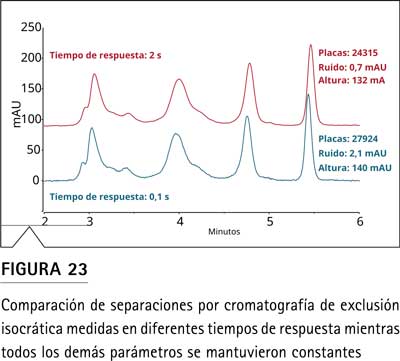
Este compromiso normalmente lo proporciona el software CDS, como el software Chromeleon CDS, que calcula los tiempos de respuesta óptimos (y la tasa de recopilación de datos) en función del ancho de pico obtenido.
Un parámetro que influye en los resultados cuantitativos relativos es el ancho de banda de, por ejemplo, un detector de matriz de diodos. El ancho de banda es el rango de longitud de onda que se utiliza para registrar el cromatograma donde la señal representa un valor de absorbancia promedio para este rango de longitud de onda.
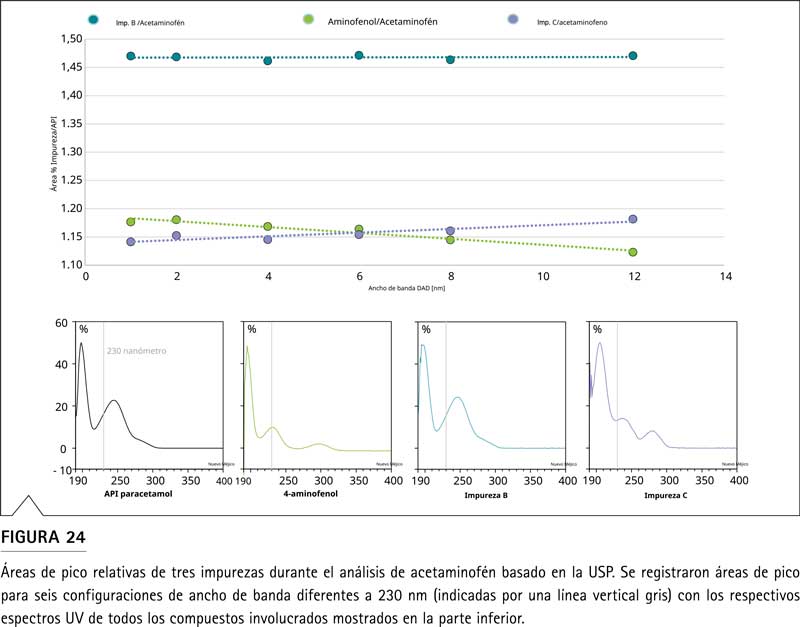
El efecto de la configuración del ancho de banda se investigó mediante un método basado en la USP que analiza el paracetamol con seis configuraciones de ancho de banda diferentes. Una primera comparación de los espectros del paracetamol y de la impureza B muestra espectros muy similares para ambos compuestos. Por lo tanto, la relación del área del pico, que a menudo se utiliza con fines de cuantificación relativa, no se ve afectada (Figura 24, línea azul). Por el contrario, los espectros de la impureza C y el 4-aminofenol tienen espectros diferentes a los del API, que se utiliza para el cálculo del área relativa del pico. Como consecuencia, la cuantificación relativa se ve afectada por la configuración del ancho de banda. Para diferentes analitos, este efecto puede incluso tener direcciones diferentes.
Mientras que para el aminofenol la respuesta relativa disminuye con un ancho de banda más amplio, el área relativa de la impureza C aumenta (Figura 24, línea verde y violeta).
Por lo tanto, recomendamos considerar con precisión la configuración del detector correspondiente durante la transferencia de un método. Cuando la transferencia se realiza en instrumentos idénticos, esto se puede hacer fácilmente. Sin embargo, cuando en la transferencia intervienen instrumentos de diferentes proveedores, se debe evaluar cuidadosamente la configuración estándar del instrumento.
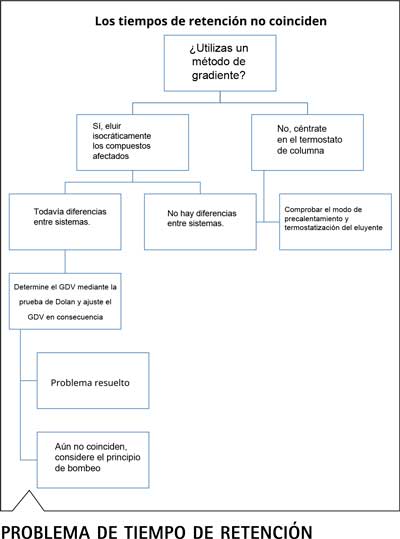
Conclusiones Problema de tiempo de retención
La transferencia de métodos de HPLC depende de varios factores diferentes que a menudo dificultan mucho esta tarea para los cromatógrafos. Por ejemplo, los tiempos de retención que no coinciden pueden deberse a:
- Diferentes principios de bombeo (bombas LPG versus HPG)
- Diferentes GDV
- Diferentes principios de termostatización de columnas
- Uso diferente del precalentador
Una pérdida de resolución también puede deberse a múltiples motivos como:
- Desajuste térmico debido al precalentamiento o termostatización de la columna
- Efectos de dispersión extra-columna adicionales
- No coinciden los disolventes de las muestras
- Configuración del detector
Estos dos criterios ilustran lo compleja que puede ser la transferencia de métodos incluso cuando solo se consideran los parámetros instrumentales: ni siquiera se tienen en cuenta los aspectos relacionados con la columna utilizada, los eluyentes u otros consumibles. Los siguientes esquemas de flujo tienen como objetivo proporcionar orientación sobre cómo transferir métodos después de ciertas observaciones. La orientación es principalmente para el análisis de la causa raíz de la desviación y no siempre para la solución final de resultados no coincidentes, lo cual se analizó en profundidad en todas las secciones anteriores.
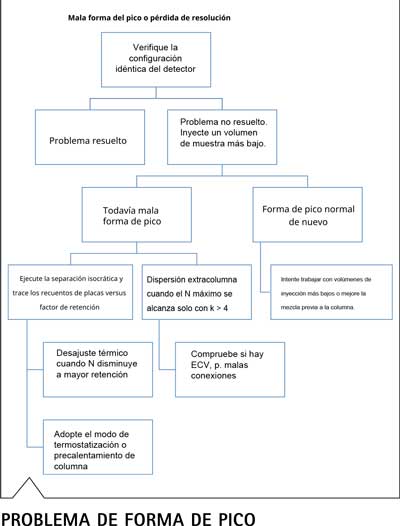
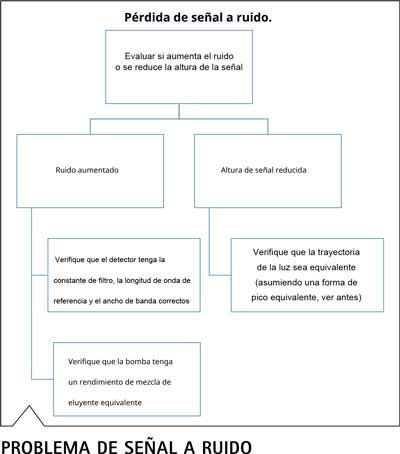
Referencias
- Guillarme, D. et al. Transferencia de métodos para cromatografía líquida rápida en análisis farmacéutico: aplicación a columnas cortas empaquetadas con partículas pequeñas. Parte I: Separación isocrática,Revista europea de farmacia y biofarmacia2007,67(3), 475-482.
- Guillarme, D. et al., Transferencia de método para cromatografía líquida rápida en análisis farmacéutico: aplicación a columnas cortas empaquetadas con partículas pequeñas. Parte II: Experimentos de gradiente,Revista europea de farmacia y biofarmacia2008,68,2, 430-440.
- Franz, H. et al., Thermo Fisher Scientific TN 75, Una herramienta universal para la transferencia de métodos de HPLC a UHPLC, 2013.https://assets.thermofisher.com/TFS-Assets/CMD/ Application-Notes/TN-75-HPLC-UHPLC-Universal-Tool-Method-Transfer-TN70828-EN. pdf
- Capítulo General de la USP <621> CROMATOGRAFÍA, Farmacopea de los Estados Unidos y Formulario Nacional,usp.org.
- Hong, P. et al., Informe técnico de Waters Corporation, Volumen de permanencia y volumen extracolumna: qué son y cómo impactan la transferencia de métodos, 2016.
- Conductividad térmica de líquidos, Ingeniería y Diseño de Transferencia de Calor, ingenierosedge.com.
- Dolan, J., Diámetro de columna, velocidad lineal y eficiencia de columna,LCGC Norteamérica,2010,28(2), 114-120.
- Manka, A. et al., Thermo Fisher Scientific TN 165, Rendimiento de detección de trazas de Bosting con el detector de matriz de diodos Vanquish y la celda de flujo LightPipe de alta sensibilidad, 2016. https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/TN-165-LC-Vanquish- DAD-Trace-Detection-TN71674-EN.pdf.




