



Método HRAM LC-MS para la determinación de impurezas de nitrosamina en fármacos
Beneficios de la aplicación
⦁ Detección y cuantificación de nueve nitrosaminas con un solo método de espectrometría de masas de alta resolución acoplado a un cromatógrafo líquido de alta presión.
⦁ Cuantificación de impurezas de nitrosamina en el principio activo ranitidina por debajo del nivel de ingesta diaria aceptable, que cumple con los requisitos de las pautas regulatorias de la FDA.
⦁ Uso del Software Chromeleon™ (CDS) Thermo Scientific™, para la recopilación y el procesamiento de datos, en un entorno totalmente compatible con 21 CFR 11, ideal para ser utilizado en instalaciones cGMP
Objetivo
Demostrar una cuantificación rápida y altamente sensible de nueve nitrosaminas con un espectrómetro de masas Thermo Scientific™ Orbitrap Exploris™ 120, y mostrar el uso del método LC-MS, para medir las impurezas de nitrosamina en productos farmacéuticos de ranitidina (Materia Prima y Producto Terminado) disponibles comercialmente
Introducción
¿Qué son las nitrosaminas?
Las nitrosaminas son sustancias químicas de pequeño peso molecular que probablemente son carcinógenos humanos, y los reguladores consideran inaceptable su presencia en medicamentos. Desde 2018, la Administración de Drogas y Alimentos de los Estados Unidos (FDA de los Estados Unidos) ha anunciado una serie de retiradas voluntarias de productos farmacéuticos tras la detección de impurezas de nitrosamina genotóxicas. Para garantizar la salud y la seguridad de los pacientes que toman estos medicamentos, las agencias reguladoras de la medicina internacional han establecido límites aceptables de ingesta diaria de nitrosaminas.1,2 Dados estos límites de acción bajos, los fabricantes de productos farmacéuticos y las organizaciones de pruebas por contrato están desarrollando métodos para cuantificar las nitrosaminas en los excipientes, los principios farmacéuticos activos (PFA) y los productos farmacéuticos para garantizar que los lotes no superen los niveles de aceptación reglamentaria y permitan el control de las impurezas dentro de sus cadenas de suministro.
La detección y cuantificación confiables de nitrosaminas, en productos farmacéuticos, a menudo son un desafío debido a la presencia de una matriz de formulación compleja y el nivel de trazas de estas impurezas en relación con la alta concentración de PFA. La espectrometría de masas basada en Orbitrap, es capaz de determinar selectivamente analitos a concentraciones bajas con alta confianza, incluso en presencia de un ruido de fondo elevado.
Varios métodos validados de la FDA de EE. UU. han demostrado una alta selectividad, sensibilidad y cuantificación de impurezas de nitrosamina en varios productos farmacéuticos.3,4,5 Dada la cantidad de productos farmacéuticos retirados del mercado y la importancia de controlar y limitar el origen de estas impurezas en los PFA, y otros productos farmacéuticos, la FDA de EE. UU. ha publicado recientemente una guía para que la industria farmacéutica exija la evaluación de riesgos, e implemente estrategias de control para limitar la formación de estas impurezas en todas las drogas humanas.6 Las nuevas pautas también sugieren expandir los controles de nitrosamina a siete impurezas, así como establecer un nuevo límite en el que, si se detecta más de una nitrosamina en los productos farmacéuticos, el contenido total de nitrosamina no puede exceder 30 ppb, o más de 26.5 ng / día. .
En esta nota describimos un método de LC-MS, recientemente desarrollado y validado, utilizando un sistema Vanquish Horizon UHPLC Thermo Scientific™ acoplado a un espectrómetro de masas Orbitrap Exploris 120, para detectar y cuantificar nueve nitrosaminas en una sola corrida analítica. También exploramos el uso de una formulación de excipiente, como una matriz alternativa a una solución ordenada para la construcción de la gráfica de calibración, y su uso para medir las impurezas de nitrosamina.
El método se realiza mediante ionización química a presión atmosférica (APCI) y puede cuantificar cómodamente las impurezas por debajo del límite recién recomendado, tanto en la solución pura como en la de los excipientes. La adquisición y el procesamiento de datos se llevaron a cabo en el software Chromeleon CDS, listo para cumplir con los requisitos regulatorios de acceso de los usuarios a los datos, la trazabilidad para las auditorías, la integridad y seguridad de los datos.
Por último, este método se aplicó para detectar y cuantificar
nitrosaminas en productos farmacéuticos con ranitidina de venta libre disponibles comercialmente. Este método también es aplicable para la determinación de nitrosaminas en otros medicamentos.
Experimental
Reactivos e insumos
⦁ Agua, grado UHPLC-MS, Thermo Scientific (P / N W81)
⦁ Metanol (MeOH), grado UHPLC-MS, Thermo Scientific(P / N A4581)
⦁ Ácido fórmico, Fisher Chemical™ Optima™ Disolvente LC / MS
⦁ (P / N A117-10X1AMP)
⦁ Clorhidrato de ranitidina, Sigma-Aldrich (R101-5G)
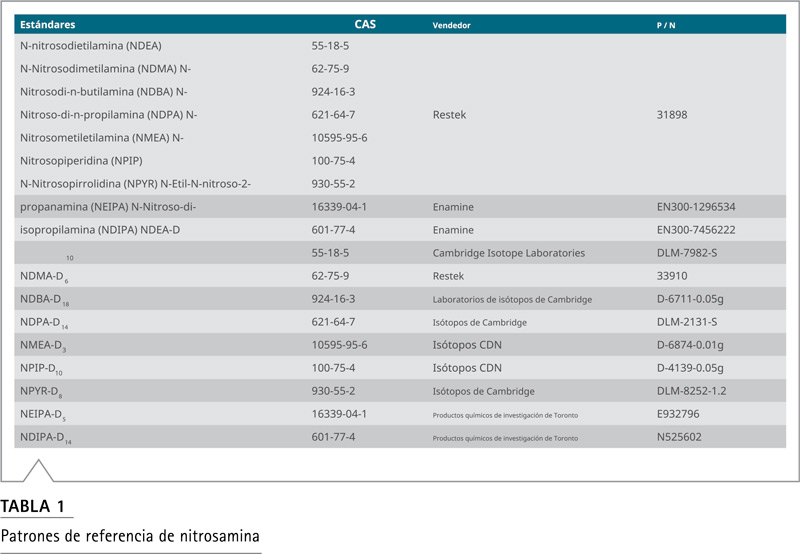
⦁ Patrones de referencia de nitrosamina (consulte la Tabla 1)
⦁ N, N-dimetilformamida (DMF), grado HPLC, Sigma- Aldrich (P / N 270547)
⦁ Tableta de 300 mg de ranitidina de venta libre
Preparación de la muestra
Preparación de la formulación del excipiente: Se preparó una matriz de formulación para imitar el ingrediente de formulación de fármaco ranitidina, mezclando 750 mg de celulosa microcristalina, 30 mg de croscarmelosa de sodio, 30 mg de estearato de magnesio, 2000 mg de hipromelosa E15 y 1000 mg de lactosa. Todos los ingredientes fueron proporcionados por nuestro socio en Patheon, en la ciudad de Bend, Oregon.
Estándares puros y excipientes: Se prepararon patrones puros combinados que variaban de 10 a 5000 ng / mL diluyendo las soluciones madre de 1 mg / mL con metanol puro. Se preparó una solución de trabajo de patrones internos combinados a 500 ng/ml diluyendo la solución madre de 1 mg / ml con metanol puro. Se prepararon estándares puros que variaban de 0,1 a 50 ng/mL mezclando 10 µL de estándares de trabajo con 10 µL de estándares internos de trabajo, seguido de la adición de 980 µL de metanol puro.
Se prepararon estándares de excipiente que variaban de 0,1 a 50 ng/mL mezclando 10 µL de solución de trabajo de estándares combinados y 10 µL de solución de trabajo de estándares internos combinados con 30 mg de mezcla de excipientes, dejándola secar a temperatura ambiente durante 30 min. y resolubilizándolo con 1 mL de metanol. La mezcla se agitó durante 40 min con un agitador mecánico, seguido de centrifugación durante 15 min a 4000 RPM. Se decantaron 750 µl de soluciones sobrenadantes y luego se filtraron usando un filtro de jeringa de PVDF de 0,2 µm.
Se preparó un estándar de referencia de DMF de 1 ppm realizando: dilución 1: 1.000.000 de DMF de calidad HPLC con metanol puro. Se inyectaron 5 µl del estándar de referencia para verificar la presencia de DMF en la sustancia farmacéutica y el producto de ranitidina comparando el tiempo de retención y los iones isotópicos controlados.
Preparación de sustancias y fármacos de ranitidina: La extracción de la sustancia ranitidina se llevó a cabo resolubilizando 30 mg de clorhidrato de ranitidina con 1 ml de metanol para obtener una concentración final de 30 mg / ml.
Se añadieron 10 µl de estándar interno de 500 ng / ml para dar una concentración final de 5 ng / ml.
Se prepararon 300 mg de extracto de tableta de ranitidina mezclando 10 ml de metanol con tableta molida para obtener una concentración final de 30 mg / ml. Se agregaron 100 μL de estándar interno de 500 ng / mL para dar una concentración final de 5 ng / mL. La mezcla se agitó durante 40 min en un agitador mecánico, seguido de centrifugación durante 15 min a 4000 RPM. Se decantaron 750 µl de soluciones sobrenadantes y luego se filtraron usando un filtro de jeringa de PVDF de 0,2 µm.
Evaluación de la reproducibilidad de la recuperación y extracción de la muestra: Para la evaluación de la recuperación de la muestra, las muestras de excipiente extraídas se prepararon agregando estándares puros a niveles de 2 ng/mL y 5 ng/mL en el excipiente en blanco antes del proceso de extracción. Para las muestras de excipientes enriquecidas, se añadieron los mismos estándares puros al excipiente en blanco después del proceso de extracción. En ambos casos, los estándares internos se agregaron después del proceso de extracción. La relación de área de pico (estándar sobre estándar interno) del excipiente extraído se comparó con la relación de área de pico del excipiente enriquecido para el cálculo del % de recuperación.
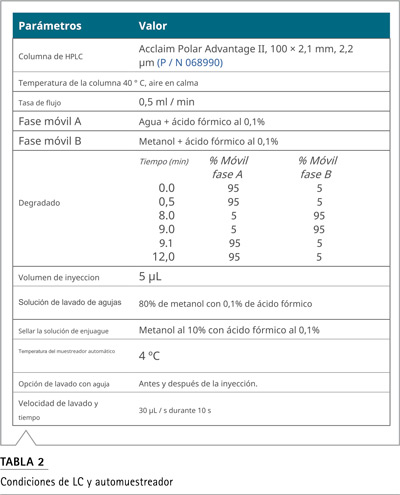
Método LC-MS
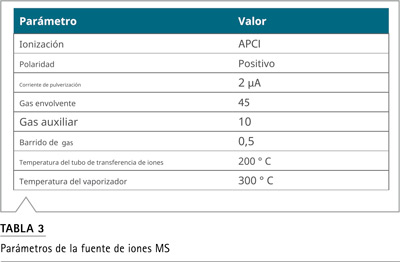
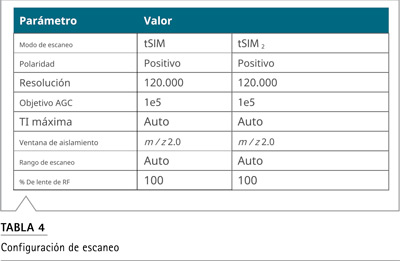
Se desarrolló un único método LC-MS dirigido, utilizando un sistema Vanquish Horizon UHPLC acoplado a un espectrómetro de masas Orbitrap Exploris 120. Se inyectaron 5 µL de muestras en una columna Aclaim Polar Advantage II, usando el gradiente de la bomba y las condiciones descritas en la Tabla 2. Todas las nitrosaminas se analizaron usando una sonda APCI con parámetros de fuente optimizados y configuraciones de exploración como se describe en las Tablas 3 y 4, respectivamente.

El software Chromeleon 7.2.10 CDS se utilizó tanto para la adquisición como para el análisis de datos para cumplir con los requisitos reglamentarios, incluido el 21 CFR Parte 11 de la FDA de EE. UU. Y el Anexo 11 de la Comisión Europea (UE).
Resultados y discusión
Determinación selectiva de impurezas de nitrosamina. El método se utilizó en la monitorización selectiva de iones dirigidaa (tSIM – por sus siglas en ingles) y la MS en tándem dirigida (tMS2) (anteriormente PRM) con una velocidad de escaneo rápida (3 Hz, con una resolución de 120.000) para una sensibilidad y selectividad óptimas de las nitrosaminas objetivo (Tabla 5).
La Figura 1 muestra una comparación del cromatograma de iones extraído (XIC) de cada impureza de nitrosamina en el excipiente en blanco con un estándar de control de 0,5 ng/ml en un ajuste de tolerancia de masa de 3 ppm. Aparte del NDBA, no se observaron otras interferencias endógenas en el extracto de excipiente usado como blanco. Aunque usando las condiciones de gradiente actuales, el método no pudo resolver a línea de base, el interferente de NDBA, la cantidad de interferente a NDBA fue <20% a 0.1 ng/mL, el calibrador más bajo e insignificante a 0.5 ng/mL. a) cuadro pag 5 B) cuadro pag 5 Figura 1. XIC de cuantificación de iones de impurezas de nitrosamina en a) excipiente en blanco yb) estándar de control de 0,5 ng / ml con un ajuste de tolerancia de masa de 3 ppm
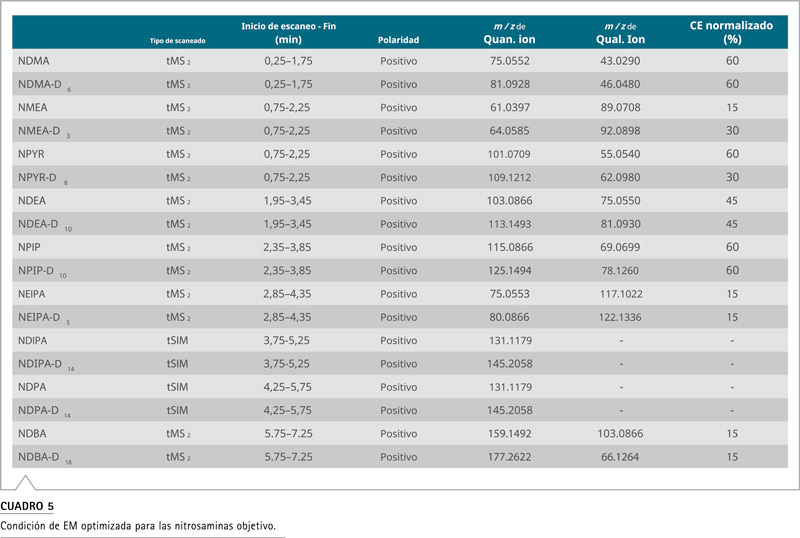
Condición de EM optimizada para las nitrosaminas objetivo

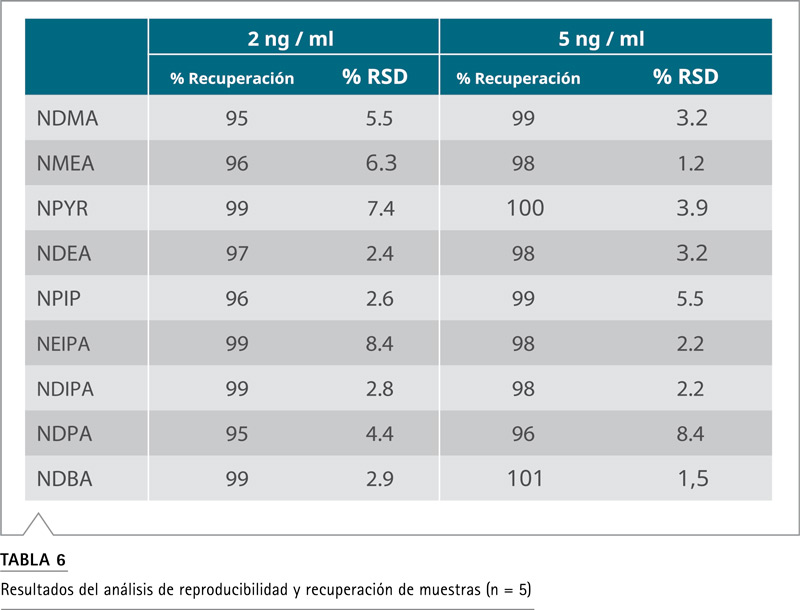
Extracción y recuperación de muestras para el análisis de nitrosaminas Un desafío crítico para detectar y cuantificar de manera confiable niveles traza de impurezas de nitrosamina en matrices complejas de formulación de medicamentos, es la necesidad de desarrollar un protocolo de extracción reproducible para maximizar la eficiencia de extracción de muestras. En respuesta a este desafío, la recuperación de la muestra y la reproducibilidad del proceso de extracción se evaluaron agregando los estándares puros de nitrosamina a niveles de 2 y 5 ng/mL en la matriz del excipiente en blanco antes y después del proceso de extracción. La Tabla 6 muestra los valores típicos de recuperación y reproducibilidad de las nitrosaminas extraídas con cinco inyecciones repetidas. La recuperación de todas las nitrosaminas durante el proceso de extracción fue entre 95 y 105%, y la reproducibilidad de las inyecciones replicadas estuvo dentro del 10% RSD.
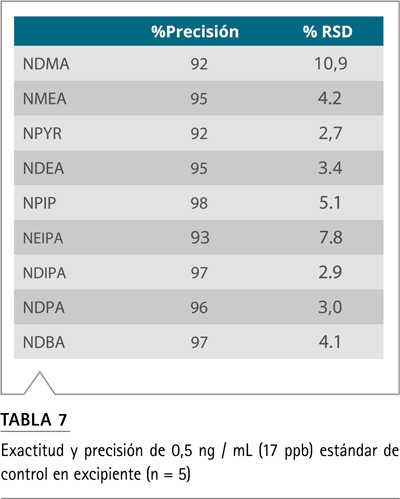
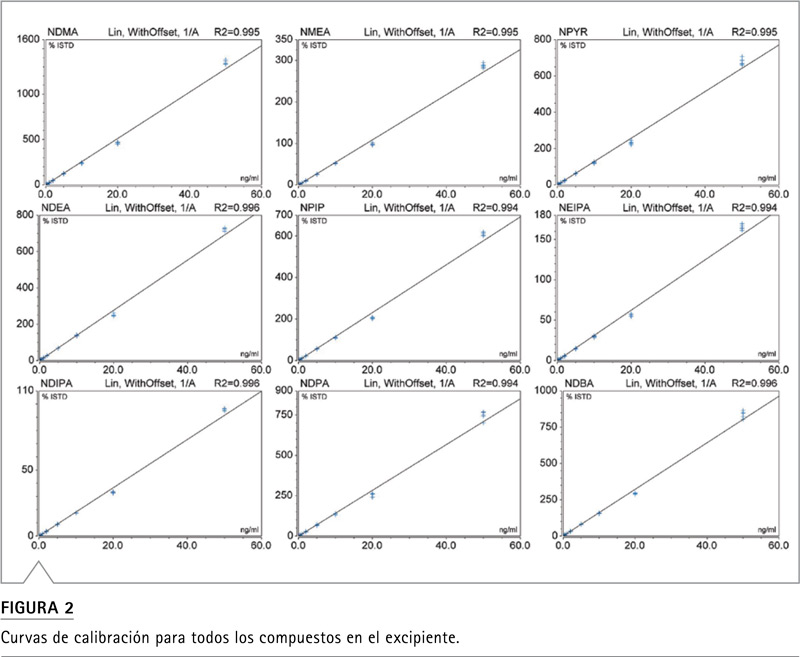
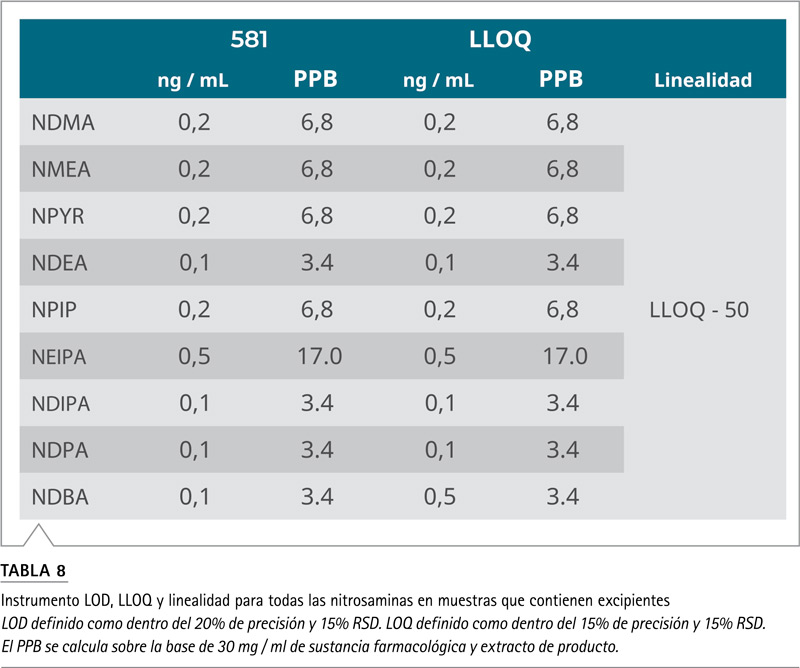
Alcanzando los límites de rendimiento reglamentarios para el análisis de nitrosaminas en productos farmacéuticos Con los nuevos límites de detección recomendados por la FDA de EE. UU. (Menos de 30 ppb para un total de 7 nitrosaminas6), es imperativo que los nuevos métodos detecten y cuantifiquen de manera precisa y reproducible las impurezas de nitrosamina en los productos farmacéuticos por debajo de ese nivel de umbral. Con las rápidas velocidades de escaneo disponibles en el espectrómetro de masas Orbitrap Exploris 120, incluso con la configuración de resolución más alta, se pudieron detectar y cuantificar todas las nitrosaminas objetivo a 17 ppb (0,5 ng / ml). La Tabla 7 muestra una exactitud y precisión típicas del estándar de control de 17 ppb en el excipiente. Las curvas de calibración se construyeron trazando las proporciones del área de los picos del estándar sobre el estándar interno frente a las concentraciones del estándar, con una ponderación de 1/x. La Figura 2 muestra la curva de calibración para todos los compuestos y la Tabla 8 enumera los límites de detección del instrumento (LOD), los límites más bajos de los límites de cuantificación (LLOQ) y los valores de linealidad para todas las nitrosaminas en el excipiente. Además de los estándares de excipientes, también se evaluaron LOD (Límite de Detección) y LLOQ (Menor límite de cuantificación), para todas las nitrosaminas en una solución pura. Los datos no se muestran aquí, pero el coeficiente de regresión lineal (R2) para todas las curvas fue superior a 0,99 y, el LOD y LLOQ resultantes para todos los compuestos, fueron casi idénticos entre las dos matrices, lo que sugiere que el método es resistente a cualquier efecto de matriz para la formulación probada.
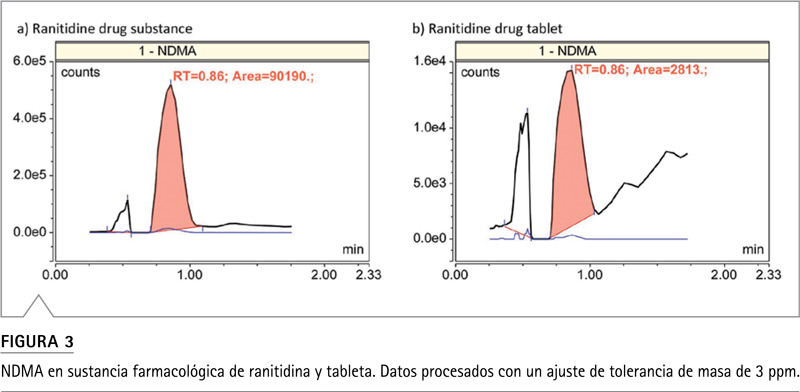
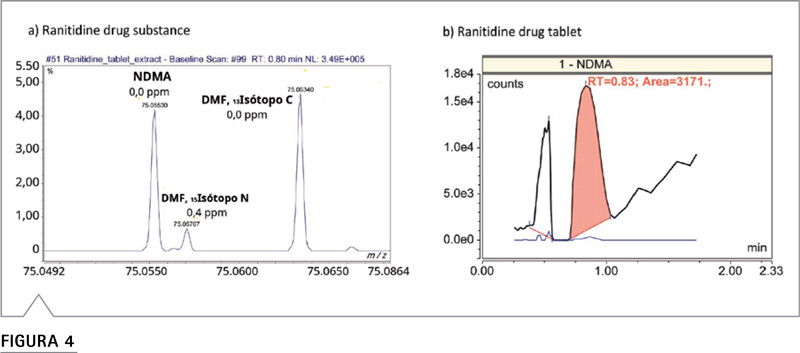
Niveles de impurezas de nitrosamina en el producto farmacéutico ranitidina Las nitrosaminas se cuantificaron en fármacos y comprimidos de ranitidina disponibles comercialmente. La cantidad medida de NDMA en la sustancia farmacéutica de ranitidina 30 mg/ml, excedió el límite superior de calibración y se estimó en más de 7 ppm (Figura 3a), mientras que la cantidad medida de NDMA en una tableta de ranitidina de 300 mg fue 82 ppb (Figura 3b), los cuales excedieron el límite regulatorio aceptable. En ambos casos, los datos se procesaron con un ajuste de tolerancia de masa de 3 ppm. La cuantificación no se vio afectada cuando se redujo el ajuste de tolerancia de masa a 1 ppm. Sin embargo, si los datos se procesaran con un ajuste de tolerancia de masa > 20 ppm (una tolerancia de masa típica que se puede lograr con espectrómetros de masas de tiempo de vuelo), esto resultaría en una sobreestimación significativa de las concentraciones de NDMA debido a la coelución con DMF. Como se muestra en la Figura 4a, el espectro de masas de NDMA en el ápice, contenía dos iones interferentes,m / z de 75.05707 y 75.06340, coincidiendo con el teórico m/z de DMF 15Isótopo N (75.05700) y 13Isótopo C (75.06340) con una precisión de masa de sub-ppm. La presencia de DMF se confirmó aún más controlando la masa monoisotópica del ion molecular (m / z 74.0600), además de la masa de los dos isótopos con una inyección del estándar de referencia DMF. El tiempo de retención resultante fue casi idéntico al tiempo de retención de NDMA en las condiciones actuales de LC (datos no mostrados). La diferencia de masa entre NDMA y DMF15El isótopo N es de solo 21 ppm, y la DMF en los productos farmacéuticos se permite hasta 880 ppm según la directriz ICH Q3C (R6). Se requiere un ajuste de resolución mínimo de 45.000 y una tolerancia de masa máxima de 15 ppm para evitar la sobreestimación de NDMA al cuantificar NDMA utilizando el ion monoisotópico. Como ilustración hipotética que se muestra en la Figura 4b, la NDMA determinada fue un 13% más alta cuando se procesó la cuantificación usando un ajuste de tolerancia de masa de 25 ppm, como se supone que es de 3 ppm (Figura 3b). Este hallazgo es consistente con el artículo de la FDA de los EE. UU. publicado recientemente en The American Association of Pharmaceutical Scientists Journal, que informa varios resultados falsos positivos para los niveles de NDMA, testeados en productos farmacéuticos de metformina, debido a la coelución de DMF, cuando el espectrómetro de masa utilizado no tiene la suficiente precisión y resolución para diferenciarlo.7
Conclusión
Se desarrolló un método rápido, altamente selectivo y sensible, utilizando la columna Acclaim Polar Advantage II, el sistema Vanquish Horizon UHPLC, el espectrómetro de masas Orbitrap Exploris 120 y el software Chromeleon CDS para la detección y cuantificación de nueve nitrosaminas en productos farmacéuticos de ranitidina disponibles comercialmente. Al combinar la cromatografía robusta y reproducible, con el poder de resolución de masa de 120.000, la velocidad de escaneo rápida y la precisión de masa por debajo de las ppm del sistema Orbitrap
Exploris 120, el método resultante puede proporcionar una cuantificación confiable y segura de nueve impurezas de nitrosamina para cumplir con las normas de septiembre de 2020 de EE. UU. de límites de aceptación regulatoria de la FDA.
Autores: Hao Yang, Thermo Fisher Scientific, San José, CA, EE. UU.
Jon Bardsley, Thermo Fisher Scientific, Hemel Hempstead, Reino Unido
Min Du, Thermo Fisher Scientific, Boston, MA, EE. UU.
Olaf Scheibner, Thermo Fisher Scientific, Dreieich, Alemania
Referencias
⦁ Conferencia Internacional sobre Armonización de Requisitos Técnicos para el Registro de Productos Farmacéuticos de Uso Humano. Directriz armonizada de ICH: Evaluación y control de impurezas (mutagénicas) reactivas al ADN en productos farmacéuticos para limitar el riesgo carcinogénico potencial. M7 (R1). 2017.http://www.ich.org/products/guidelines/multidisciplinary/article/ multidisciplinary-Guidelines.html
⦁ EMA / 369136/2020 Procedimiento de conformidad con el artículo 5, apartado 3, del Reglamento CE (nº) 726/2004 Impurezas de nitrosamina en medicamentos de uso humano. https://www.ema.europa.eu/en/ documents / referral / nitrosamines-emea-h-a53-1490-assessment-report_en.pdf
⦁ Método de cromatografía líquida-espectrometría de masas de alta resolución (LC-HRMS) de la FDA de EE. UU. FY19-177-DPA-S para la determinación de NDMA en sustancias farmacéuticas y productos farmacéuticosranitidina.https://www.fda.gov/media/130801/download
⦁ US FDA FY19-107-DPA-S, Método de cromatografía líquida-espectrometría de masas de alta resolución (LC-HRMS) para la detección de seis impurezas nitroso en ARB. https://www.fda.gov/ media / 125478 / download
⦁ Método de cromatografía líquida-ionización por electropulverización- espectrometría de masas de alta resolución (LC-ESI-HRMS) de la FDA de EE. https: // www. fda.gov/media/138617/download
⦁ US FDA-2020-D-1530, Control de impurezas de nitrosamina en medicamentos humanos, Guía para la industria. https://www.fda.gov/media/141720/download
⦁ Yang, et al; Una advertencia: Procedimientos analíticos cuantitativos de LC-HRMS para el análisis de N-nitrosodimetilamina en metformina;La revista AAPS, 2020, 22:89.
Descubra más en thermofisher.com/nitrosamines
Más información:
info@sol-analiticas.com
www.sol-analiticas.com
Conozca más sobre espectrofotometría







